

时间:

作者:保敏敏、吕蓓蓓、魏文芝、张敏娟
目的 建立往复筒溶出方法考察奥卡西平刻痕片仿制药与原研药分割后半片制剂的体外溶出行为相似性,评价原研药和仿制药的分剂量药学特性差异。
方法 以pH1.2盐酸溶液、pH4.5醋酸盐缓冲液、pH6.8磷酸盐缓冲液和水(均含0.5%十二烷基硫酸钠 250 mL)为溶出介质,往复频率为10dip·min–1,采用往复筒溶出装置测定仿制药和原研药的溶出曲线,结合相似因子(f2)法评价仿制药和原研药的溶出行为相似性,并与桨法进行比较。采用脆碎度检测仪及电子天平,通过人工掰分法和切药器法测定各厂家半片制剂的脆碎度、分割后质量差异及质量损失。
结果 仿制药A在4种溶出介质中的 f2 均>50,与原研药的溶出行为相似;仿制药B在4种溶出介质中f2均<50,和原研药的溶出行为不相似。仿制药A、B分割后质量差异、质量损失和脆碎度均高于原研药。
结论 采用往复筒法测定奥卡西平半片制剂的溶出曲线相较于桨法具有良好的区分力,且仿制药的分剂量质量控制相较于原研药仍有一定差距。

奥卡西平是一种新型抗癫痫药物,可用于治疗成人和儿童癫痫的简单或复杂部分性发作、全身强直阵挛发作等,酶诱导作用弱,不良反应少。目前,奥卡西平上市剂型主要为刻痕片,可掰分后使用,便于灵活调整剂量,方便用药人群,降低治疗成本。刻痕片的大小、硬度和刻痕线的形状、深浅等因素会对分剂量的准确性造成影响,进而出现剂量偏小致使疗效减弱或偏大而引起不良反应的问题。尤其对于老年人、儿童等特殊患者群体和治疗窗窄的药物,需要格外关注分剂量用药的安全性。目前国内对于刻痕片多探讨该剂型在临床使用时分剂量的均匀性,相关深入评价研究较少,对于内在质量参数考察不足,中国药典 2020年版尚未收录相关通则。
往复筒法作为近年得到较快发展的新型溶出度检查方法,已被各国药典收载为标准方法,但国内相关实验研究较少。该装置主要通过将样品放置在玻璃往复筒中,控制往复筒在溶出杯中的往复运动来测定药物溶出量,具有更接近人体胃肠道蠕动生理环境的运行条件,能够良好地反映体内外相关性。研究表明往复筒法对于部分普通片剂能够实现和篮法或桨法类似的流体动力学特点,提供区分力良好的溶出曲线,具有评价溶出行为一致性的潜在优势。
目前各国药典标准所收载奥卡西平片溶出度检查方法均为桨法,以含 0.6%十二烷基硫酸钠(sodium dodecyl sulfate,SDS)水溶液作为溶出介质。本研究采用往复筒法测定奥卡西平半片制剂的溶出曲线,并与桨法进行比较,得到更具有区分力的溶出曲线,考察进口原研药与国产仿制药半片制剂的溶出行为差异。同时根据美国食品药品监督管理局(FDA)及国家药品监督管理总局药品审评中心发布的仿制药功能性刻痕相关研究指导原则,对奥卡西平片进行分割后质量差异、质量损失、脆碎度参数的考察,为国内奥卡西平片仿制药一致性评价研究工作和处方工艺开发提供参考。
2.1 仪器
HPLC仪(Agilent1260型,G7111A型四元泵,G7129A 型自动进样器,G7116A 型柱温箱,G71157型二极管阵列检测器)、往复筒溶出仪(推荐使用华溶DS-BIOAT往复筒法溶出系统);真空脱气仪(推荐使用华溶DGU-900在线溶媒脱气机);PB-21型酸度计、CP225D 型十万分之一电子天平均购自德国Sartorious 公司;FT-2000型脆碎度检测仪(天津市矽新科技有限公司);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司)。
2.2 试药与试剂
奥卡西平对照品(中国食品药品检定研究院,批号:100657-201102;纯度:99.8%);奥卡西平片原研制剂(瑞士Novartis公司,规格:300mg;批号:TT434、TV182、TU897);奥卡西平片仿制制剂(国内A企业,规格:300mg;批号:18108、19003、19410,仿制药 A),奥卡西平片仿制制剂(国内B企业,规格:300mg;批号:11805、11057、19102,仿制药 B);SDS(SIGMA-ALDRICH,批号:SLBZ9853;含量≥99.0%);SDS(福晨化学试剂有限公司,批号:20190220;含量≥59.0%);SDS(国药集团化学试剂有限公司,批号:F20090326;含量≥86.0%);乙腈为色谱纯,其余试剂均为分析纯,水为超纯水。
3.1 色谱条件
色谱柱:ZORBAX SB C18(150mm×4.6mm , 5 μm);流动相:乙腈-0.05mol·L–1磷酸二氢钾溶液(含0.2%三乙胺,用磷酸调节 pH至6.0)(35∶65);检测波长:256nm ;流速:1.0mL·min–1;柱温:30 ℃; 进样量:10μL。
3.2 溶液的制备
3.2.1 对照品溶液的制备
精密称取奥卡西平对照品约50mg,置50mL量瓶中,加流动相溶解并稀释至刻度,作为对照品储备液,精密量取2mL, 置10mL量瓶中,用流动相溶解并稀释至刻度,摇匀,制成浓度约为 200μg·mL–1的对照品溶液。
3.2.2 供试品储备液的制备
精密称取奥卡西平片 ( 批号 :18108)细粉适量(约相当于奥卡西平50mg),置50mL量瓶中,加流动相溶解并稀释成每1mL约含1mg奥卡西平的供试品储备液。
3.2.3 供试品溶液的制备
精密称取奥卡西平片 ( 批号:18108) 细粉适量 (约相当于奥卡西平10mg),置50mL量瓶中,加流动相适量,超声使奥卡西平溶解并定量稀释至刻度, 摇匀,滤过,取续滤液,即得。
3.2.4 阴性样品溶液的制备
按奥卡西平片处方比例制成缺奥卡西平的空白辅料样品,称取适量,加流动相溶解,定量稀释并滤过,得到阴性样品溶液。
3.3 方法学验证
3.3.1 专属性试验
分别取“2.2”项下对照品溶 液、供试品溶液和阴性样品溶液于“2.1”项下色谱条件进样测定,记录色谱图,见图1,结果表明辅料对主峰测定无干扰。
3.3.2 线性关系考察
分别精密移取“2.2.1”项下 对照品储备液 0.5 ,1,2,4,8 mL ,置10 mL量瓶中,用流动相稀释至刻度,摇匀,取上述溶液及对 照品储备液按“2.1”项下色谱条件进行测定。以色谱峰面积(Y)与对应的质量浓度(X)进行线性回归,得回归方程 Y=16 349X+64.506(r=0.999 9)。结果表明奥卡西平在 49.9~998.0 μg·mL–1 内与峰面积之间 线性关系良好。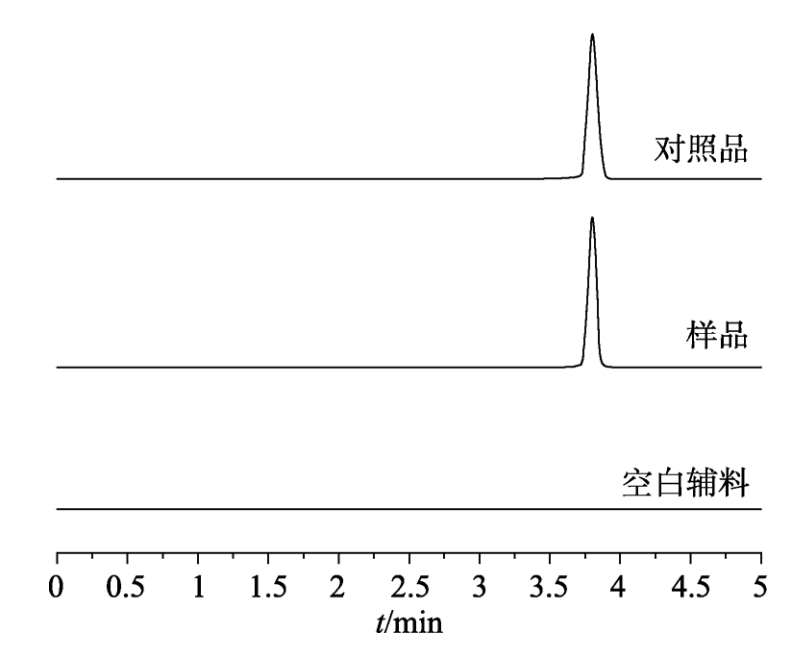
3.3.3 仪器精密度试验
取“2.2.1”项下奥卡西 平对照品溶液,按“2.1”项下色谱条件连续进样 6 次, 记录峰面积。结果显示RSD为 0.1%(n=6),本方法仪器精密度良好。
取“2.2.1”项下奥卡西 平对照品溶液,按“2.1”项下色谱条件连续进样 6 次, 记录峰面积。结果显示RSD为 0.1%(n=6),本方法仪器精密度良好。
3.3.4 重复性试验
取同一批奥卡西平片(批号:18108),按“2.2.3”项下方法平行制备供试品溶液6 份,按“2.1”项下色谱条件进行含量测定。测得奥卡西平含量的 RSD为1.2%(n=6),表明本方法重复性良好。
3.3.5 加样回收率试验
移取“2.2.2”项下供试 品储备液2mL ,置10mL 量瓶中,共12份,再分别精密加入“2.2.1 ”项下对照品储备液 0.4,2.8,4,5.2mL,各3份,用流动相稀释至刻度,摇匀。按“2.1”项下色谱条件进样测定,外标法计算回收率,结果见表1 ,表明方法回收率良好。
3.3.6 溶液稳定性试验
取“2.2”项下对照品溶 液和供试品溶液, 室温放置 0,2,6,12,18,24 h,按“2.1”项下色谱条件测定,记录峰面积。奥卡 西平对照品和供试品峰面积的RSD分别为0.4%, 0.5%,表明该溶液稳定性良好。
3.3.7 耐用性试验
取“2.2.3”项下供试品溶液, 在不同pH(5.8,6.0,6.2)、不同柱温(25,30,35 ℃)以及不同流速(0.8,1.0,1.2mL·min–1)条件下按“2.1”项下色谱条件进样测定。结果显示pH、柱 温、流速于一定范围内变化时, 奥卡西平含量的RSD 均<2%,对测定结果无影响,表明该方法耐用性良好。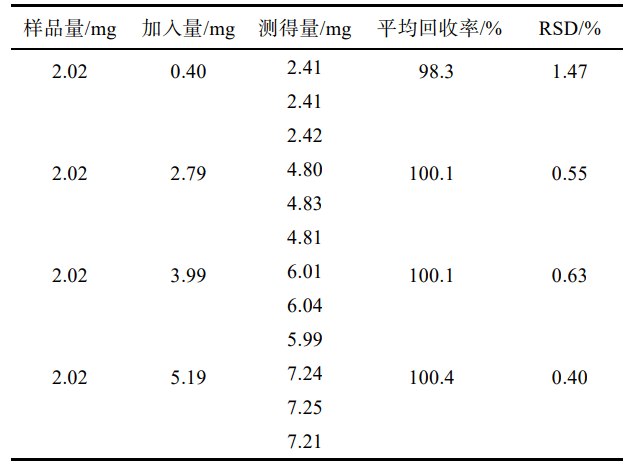
3.4 往复筒溶出方法的确定
3.4.1 SDS 浓度的考察
以水为溶出介质,分别测 定加入 SDS 0.3%,0.4%,0.5%和 0.6%时原研药和 仿制药的溶出度,比较不同SDS用量对奥卡西平半片的溶出影响,见表2。结果显示,加入0.3%,0.4%SDS 时各厂家制剂累积释放量均<80%。加入0.6%SDS时 2 种仿制药的相似因子均>50,溶出条件宽松,无区分力。因此确定SDS的用量为0.5%。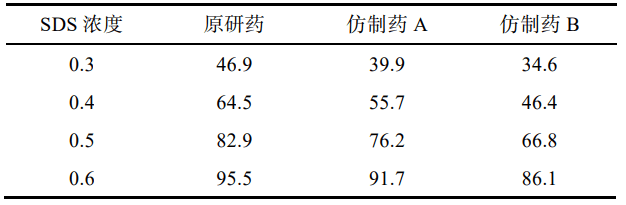
3.4.2 往复频率的考察
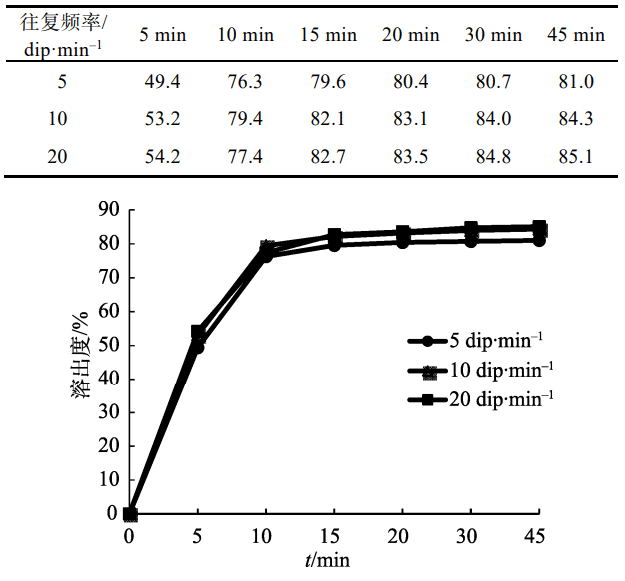
以0.5%SDS水溶液250 mL为溶出介质,分别测定原研药半片在往复速率为 5,10,20dip·min–1时的溶出度,结果见表3。不同往复频率下原研药的溶出度无显著差异,溶出曲线基本一致,见图2。往复频率为10dip·min–1时起泡较少,故本研究选用10dip·min–1进行溶出度的测定。
3.4.3 SDS 来源的考察
选取进口以及2个国产厂家生产的SDS,以10dip·min–1测定原研药在 0.5%SDS 水溶液中的溶出度,分别于5,10,15,20,30,45min取样1.5mL,按“2.1 ”项下色谱条件测定。
结果显示,使用SIGMA- ALDRICH、福晨和国药生产的SDS得到的样品10 min 时的溶出度RSD分别为2.1%,11.8%和7.5%。SIGMA-ALDRICH生产的SDS测得的样品批内均一性良好,选择该厂家SDS作为溶出度试验的增溶剂。见表4。

3.5 溶出曲线测定
3.5.1桨法
参照美国药典(USP43)中对于150mg 规格的奥卡西平片采用的溶出度测定方法,取本品6 片,以掰分后半片作为溶出样品,转速60r·min–1,温度37℃,溶出体积900mL,分别在含0.3%SDS 的 pH1.2 盐酸溶液、pH4.5醋酸盐缓冲液、pH6.8磷酸盐缓冲液和水中测定 溶出度,于5 ,10,15,20,30,45min时取溶液适量,0.45μm微孔滤膜滤过,作为供试品溶液。另精密称定奥卡西平对照品适量,加流动相定量稀释成质量浓度约为200μg·mL–1的溶液,作为对 照品溶液进样。按“2.1”项下色谱条件测定,外标法计算不同时间点各厂家半片的溶出量,绘制溶出曲线,结果见图3。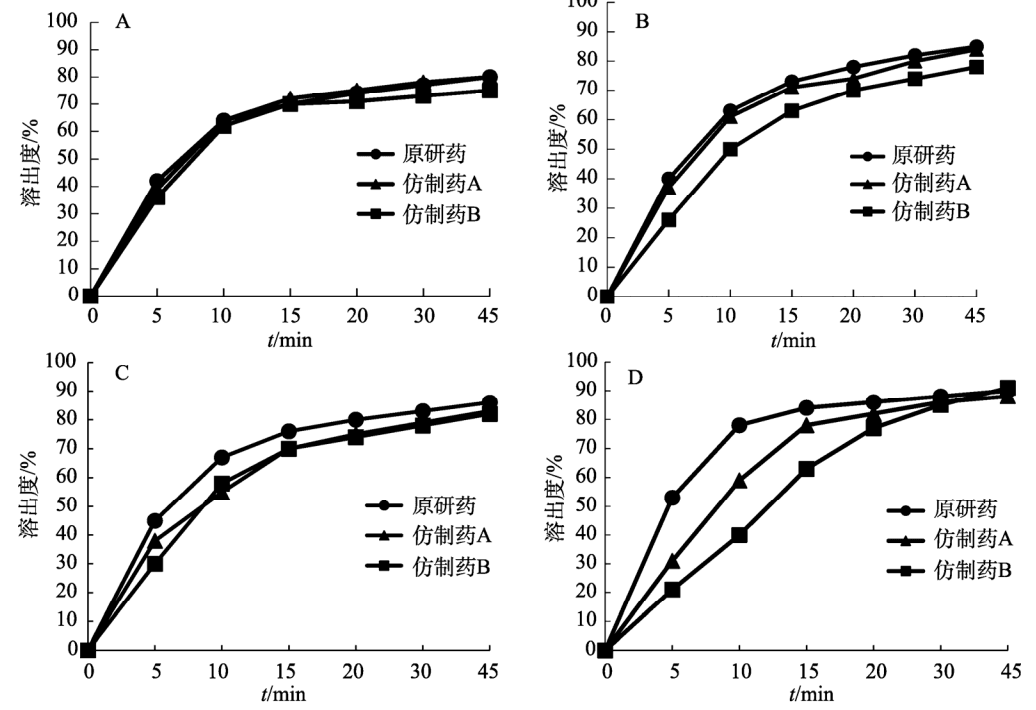
3.5.2 往复筒法
取本品6片,以掰分后半片作为溶出样品,采用往复筒溶出装置的单排管进行溶出试验,设定往复速率10dip·min–1;不加上筛网,下筛网选择100目,材质为聚丙烯酰胺;水浴温度37 ℃ ; 分别以 0.5% SDS水溶液、0.5% SDS盐酸溶液 (pH1.2) 、 0.5% SDS醋酸盐缓冲液(pH 4.5) 、0.5%SDS 磷酸盐缓冲液(pH 6.8)为溶出介质,体积为250mL;分别在 5,10,15,20,30,45min 时取样1.5mL,用 0.45μm微孔滤膜滤过,作为供试品溶液。另精密称定奥卡西平对照品适量,加流动相定量稀释成质量浓度约为600μg·mL–1的溶液,作为对照品溶液进样。按 “2.1”项下色谱条件测定,外标法计算不同时间点各厂家半片的溶出量, 绘制溶出曲线,结果见图4。
3.6 相似性评价
根据《普通口服制剂溶出度试验技术指导原则》,通过计算原研制剂与仿制制剂在不同溶出介质中的相似因子(f2)评价溶出曲线相似性,结果见表5~6。往复筒法相较于桨法,所得到的溶出曲线 具有更强的区分力。
3.7 质量差异

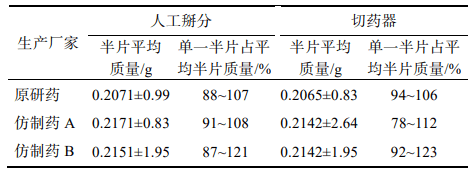
随机选取本品30 片,以人工掰分和切药器2种不同的方式分割成2个半片, 取每片中1个分割后部分进行称重,比较其与平均半片的质量差异。分割后部分质量超出平均半片质量85%~115%的不能>1个,如果有>1个分割部分超过上述标准,或者有1个分割部分>75%~125%的限度,则不符合要求。结果显示,原研药通过2种分割方法得出的半片质量均一性均良好,未出现不合格半片;仿制药A经切药器法得到的半片质量差异大且出现1个不合格半片;仿制药 B人工掰分和切药器法各出现1个不合格半片。半片质量计算结果见表 7。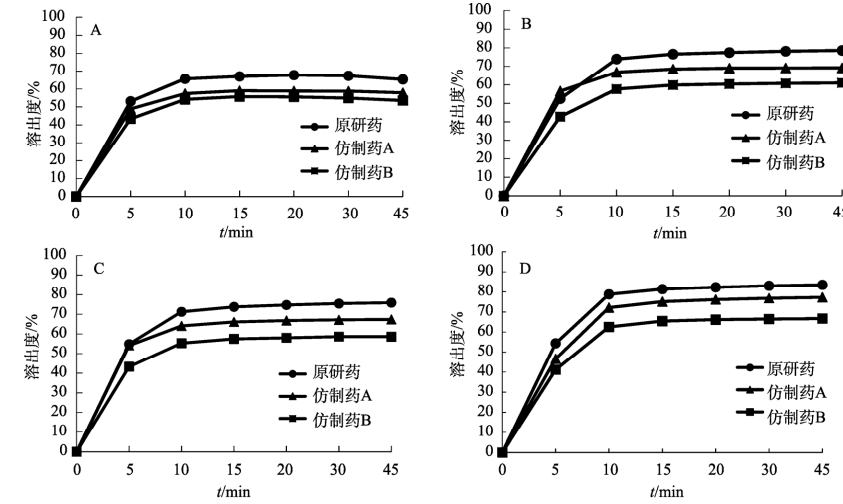
3.8 分割质量损失
用手掰和切药器法各自分割15整片并精密称定,和分割前质量比较,计算分割后整片的质量损失。结果显示,原研药的分割质量损失低于仿制药,仿制药A在切药器法中质量损失最大,仿制药B通过人工掰分质量损失最多,见表 8。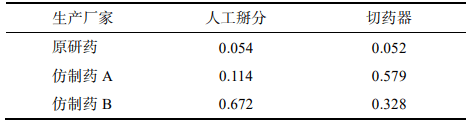
3.9 脆碎度
根据中国药典2020年版通则0923,采用脆碎度检测仪对原研药和仿制药分割后半片进行检查,质量减失均<1%,且无断裂、龟裂及粉碎片。2 种分割方式下原研药的脆碎度均低于仿制药。仿制药B通过人工掰分法得到的半片脆碎度最高。见表 9。
4.1 色谱条件的选择
采用中国药典 2020年版奥卡西平片含量测定 方法时发现主成分峰前出现一个分离度差的色谱峰,影响含量测定的准确性,经与对照品色谱图比较,排除其为杂质峰的可能。考虑奥卡西平结构中存在酰胺基,pH3.0的流动相可提供H+ ,酸 催化条件下奥卡西平易发生酰胺-亚胺醇式异构化反应,出现2种异构体的动态平衡,可能是导致该 HPLC 方法色谱图出现无法分离的两色谱峰的原因。参考美国药典(USP43)方法将pH调至6.0,并测试不同品牌的C18柱系统适用性,结果显示奥卡西平主峰前后均未出现分裂峰,主峰峰形良好。因此本研究采用调整后的色谱方法进行奥卡西平半片的含量测定。
4.2 溶出装置的选择
本研究采用往复筒法,在4种溶出介质中均得到比桨法更有区分力的溶出曲线,结合相似因子说明仿制药A与B之间、仿制药与原研药之间的溶出行为均存在显著性差异。采用该方法可以进一步评价产品内在品质,从而为改进仿制药生产工艺提供数据参考。
4.3 筛网规格
往复筒溶出装置的上下筛网规格一般为8~400 目,本研究通过试验不同目数的筛网发现上筛网添加与否对药物在往复筒中的溶解无明显影响。对下筛网选用40,78, 100目进行考察,发现目数为100目时能够防止样品粉末漏出往复筒,避免溶出损失,故最终确定不加上筛网,下筛网为100目,可满足实验要求。
4.4 可分割性差异
仿制药A的外观和原研药类似,为浅黄色胶囊形片, 双面中刻痕。在使用切药器时,由于其质地比较坚硬,切割阻力大易导致不规则崩裂现象,继而出现分割后半片不均一、分割质量损失高的情况。仿制药B为圆形片,单面中刻痕且刻痕较浅。其片形使得徒手掰分比较费力,容易出 现质量不均一的半片,导致分割后片剂的质量损失较仿制药A更为显著。因此,仿制药的刻痕功能在分割剂量方面不及原研药。
本研究采用往复筒法比较国产与进口奥卡西 平半片制剂的体外溶出行为,在4种溶出介质中获得的溶出曲线均能有效区分不同厂家的奥卡西 平片。结果显示国产仿制药的溶出行为与进口原研药有不同程度的差异。同时通过考察分割质量损失等刻痕片的关键质量属性,提示国产仿制制 剂分剂量质量控制存在提升空间。
略
如需原文,请联系小编






