

时间:

胃肠道(GI)是最受欢迎使用的给药途径之一,因为与其他途径相比,胃肠道可以提高患者的依从性并降低成本。然而,在开发更复杂的剂型时,如结合两种或多种药物的剂型,其复杂的性质带来了巨大的挑战。固定剂量复方制剂(FDC)是两种或多种单一活性成分组合在一个单一剂型中。这种处方组成代表了一种新的处方,与单独的每种单一产品相比,它是安全有效的。通过复杂途径给药的复杂药物产品,需要对配方开发进行深入的考虑。此外,在证明此类仿制药的生物等效性(BE)时,从监管角度来看,这是一个挑战。本报告向读者简要介绍了2018年11月3日和4日在华盛顿举行的药物科学家年度协会(AAPS)会议上进行的为期两天的短期课程。这篇手稿将全面了解胃肠道生理学对药物吸收最有影响的方面,以及目前的技术,以帮助理解口服药物产品在以胃肠道为代表的复杂环境中的过程。通过对FDC产品开发和监管问题的案例研究,本文将为读者提供一个探索开发FDC产品的常规方法。
从解剖学的角度来看,胃分为胃底、胃体和胃窦,但就运动功能而言,可以分为两部分:由胃底和胃体近端组成的近端胃和由胃体远端和胃窦组成的远端胃。近端胃的运动特征是平滑肌(张力)保持收缩状态,而远端胃产生阶段性收缩。在消化间期,胃近端肌张力高,而远端胃处于一种周期性运动模式,称为迁移运动复合体(MMC)。这种复合体涉及胃和大部分小肠(但不包括远端小肠),分为三个阶段:第一阶段,无收缩的静止期;第二阶段,随机收缩;第三阶段,突然出现的重复性收缩,也会突然结束;第三阶段可以在胃或小肠近端开始,然后向回肠远端迁移。胃pH在MMC期间波动,胃窦pH在第三阶段收缩开始前最低(酸性更强),在第一阶段开始时较高。这种pH值的变化是由于MMC的第三阶段以及十二指肠的无胆汁碳酸氢盐回流导致的酸和胃蛋白酶分泌增加。肠道和胰腺分泌物(如水、碳酸氢盐和胰酶)在小肠第三期收缩期间增加。一旦食物被摄入,近端胃就会放松以容纳食物,然后近端胃就会紧张性收缩,将食物推向更远的地方。远端胃会通过有力而有规律的收缩来混合和研磨食物。十二指肠在摄入食物后几乎直接暴露于营养物质中,这将激活多种十二指肠负反馈机制,例如通过迷走神经反射和激素信号介导的负反馈机制。这将通过抑制近端胃张力和收缩,刺激幽门闭合,从而延迟酸性、高渗或高热量胃内容物进入十二指肠。食物的物理状态一致性、脂肪含量和热量负荷在调节胃的运动反应中起着相关的作用。低热量的液体在由胃底张力和远端胃的少量运动以指数方式产生的压力梯度下排空。更黏稠的食物需要经胃窦研磨,直至粒度减小。胃减少颗粒所需的时间可以解释排空开始前观察到的所需时间。因此,胃排空分为两个阶段:缓慢排空期(负责固体物质的消化)和排空期即消化后的固体颗粒或液体可以很容易地从胃中排空的线性排空期。不可消化的固体通常在MMC的消化I第三阶段从胃中排空。最近的一项研究证明了这些第三阶段收缩对将布洛芬从胃排空到小肠产生影响。这些收缩在肠道吸收中pH起着关键作用,决定了Cmax和Tmax。最近进行了一项临床排空研究,以研究禁食和进食状态下一杯水的胃排空率。将标准剂量的酚红添加到一杯水中,并由健康受试者摄入。喝了这杯水后,从胃、十二指肠和空肠抽出胃肠液。基于计算模型,作者发现,一杯水的胃排空非常迅速,尤其是在禁食状态下。
闪烁扫描成像被认为是研究人类胃排空的金标准,通常由1小时、2小时和4小时的胃滞留百分比来定义。然而,使用单一的测量结果总结并不能获得上述由饮食激活的复杂机制的报告。正如Parker及其同事所描述的,诺丁汉大学已经验证并公布了基于液体餐的胃排空测试的正常值,以获得对胃运动和感知功能的全面评估。该测试可以区分液体餐时的胃排空的早期和晚期,这可以反映胃的调节和胃窦的排空。近年来发展了两种研究胃功能的技术。SmartPill®是一种可摄入的设备(26mm × 13mm),可测量腔内pH值、压力和温度。它将数据无线传输到一个可穿戴的外部记录器,允许在家进行动态研究。肠道pH值的变化,以及排便后温度的下降,可以精确测量区域和整个肠道的转运时间。然而,需要注意的是,考虑到该装置的尺寸并不能反映正常可消化食物的胃排空情况,而且确实发现胃排空时间比闪烁成像测量的时间更长。无论如何,该技术具有非侵入性以及结合整个肠道转运过程pH值测量的优点。除了考察胶囊外,磁共振成像(MRI)最近也被应用于胃肠道功能的研究,与其他技术相比,这项技术具有一些主要优势:它是非侵入性的,不会使受试者暴露于辐射中,并且不需要任何造影剂。这是一种可以同时测量胃、小肠和结肠的体积、物理化学特征以及转运速率,并量化运动方式的独特的技术。这项技术尚未在各研究中心建立标准化,目前还不允许评估直立状态下的胃功能。总之,人类的胃看起来更复杂,并且在体内药物行为以及肠道吸收中发挥主要作用。
除了胃,小肠在药物吸收方面发挥关键作用。特别关注(i)肠液体积,(ii)肠液的特征和组成,以及(iii)肠壁对药物化合物的渗透性。肠道内的残液量相当少,而且不像一池水一样从近端向远端均匀分布。这些液体合理的分布在不同的流体库中。正如Mudie及其同事所强调的那样,健康受试者的储库数量和每个储库的实际体积差异非常大。这一发现是一项重要的研究,让配方科学家意识到肠道不像一个“游泳池”,完全充满了水。根据Mudie等人的观察,使用流体动力学而不是静态和大体积来预测口服药物的体内特性更为准确。这是在泊沙康唑(一种弱碱性化合物)中观察到的,通过使用模拟软件中的动态流体和pH模型来预测其体内性能。尽管该模型对预测水溶性较差的化合物的体内性能有影响,但作者得出结论,该模型对预测溶解度较高的化合物的全身暴露可能没有很大影响。此外,如前所述,在储库的数量和体积上存在巨大的差异。例如,一名受试者显示只有2个储库,总容积为1.4 mL,而另一名受试者显示有23个储库,总容积为160 mL。一项后续研究旨在揭示液体库的出现与当前运动特性之间的潜在联系。20世纪70年代,Vantrappen等人在上消化道第三阶段收缩后不久观察到碳酸氢盐的分泌率较高。这样,进入小肠的胃酸可以直接被碳酸氢盐中和。这种所谓的促分泌物很可能是肠道内形成液体储液库的一个重要因素。除了获得有关胃肠道中体积的知识外,这些流体的组成是另一个重要方面。在最近的一项研究中,从20名处于禁食和进食状态的健康受试者身上抽取人类十二指肠液。通过摄入液体膳食(即400 mL的Essure Plus®,等于700卡路里)来模拟进食状态。在获取这些液体作为时间的函数后,分析液体的pH值和内源性成分(胆汁盐、磷脂、胆固醇、酶活性和脂质消化产物)。这项研究的结果表明,这些成分的存在因人而异,尽管每个人的研究方案都是相同的。特别是对于离子化的化合物,肠道中的pH值对于药物的溶解和随后的吸收是极其重要的。Amidon教授(密歇根大学)的研究小组在禁食和进食状态下口服布洛芬缓释片(800 mg)后,从37名健康受试者身上吸出胃肠道液体。从胃肠道的不同部分吸出液体:胃、十二指肠和空肠。本研究显示,pH值的高度波动,特别是在十二指肠,这是除运动外的一个重要内在因素,解释了受试者之间和受试者内部布洛芬全身暴露的差异(图1)。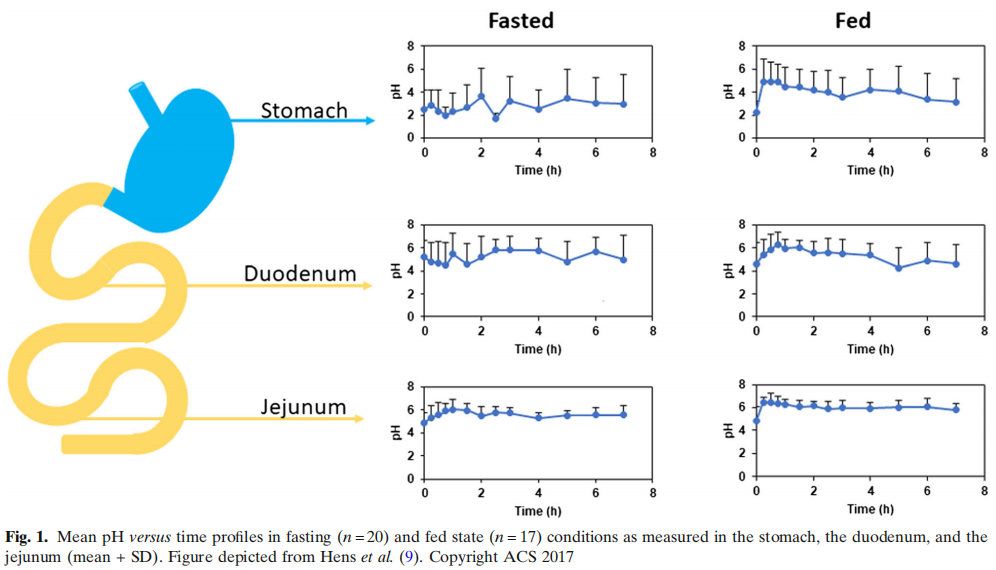
除了溶解度外,吸收一直是评估药物性能的关键参数。文献中描述了多种技术来评估药物化合物的肠道通透性。文献中描述了多种技术来评估药物化合物的肠道渗透性。Loc-l-Gut®方法,即双球囊灌注系统,是一种有趣的研究技术,用于探索药物化合物在胃肠道不同区域的渗透性。胃肠道的特定区域将被两个球体所膨胀,从而分离出基于性质的特定区域。随后,将灌流药物溶液,消失的药物量是衡量药物吸收量的指标。这项技术的应用已经揭示了氢化可的松在十二指肠、空肠和回肠的肠道渗透性。Dahlgren等人最近的一篇综述汇编了在人类肠道所有部位进行的61项研究中对80种物质进行的273次单独测量的历史Peff数据。这一令人印象深刻的数据为研究人员提供了参考,以优化体外研究的方案,从而提高其内部吸收的预测。
关于结肠生理学,应用高分辨率测压法(HRM)的最新发现表明,肠运动主要表现为非传递和逆行向后运动,并且这两种活动在进食后不久都会增加。这些结肠运动模式具有延迟结肠内容物到达直肠的作用,并有利于横结肠和升结肠的充盈,而横结肠和升结肠通常是传播性收缩开始的地方。传播性收缩,包括带有固体内容物结肠运动相关的高振幅传播性收缩代表了结肠活动的一小部分,通常在饭后约1-2小时和醒来时更频繁。其原因很可能与以下事实有关:在白天的这些时刻,夜间和消化间期积聚在远端小肠的内容物的到达决定了升结肠和横结肠的膨胀,从而触发了传播活动。非传播性运动的普遍存在解释了与小肠相比,正常结肠运输时间较慢(约35小时)的事实。这使结肠能够发挥其吸收和发酵的功能,并成为一个足够大的储存器官。HRM是研究结肠运动功能的一种有用技术,但具有侵入性,通常需要肠道特殊准备。这使得当需要在生理条件下研究结肠功能时,该技术的吸引力降低。最近,其他技术已被应用于结肠功能的研究。电磁胶囊是一种可摄入的涂有硅胶的圆柱形磁体(21毫米x 8毫米),用于绘制结肠内容物的实时运动图。一个装有4x4磁场传感器检测平板佩戴在患者腹部周围,以监测药物的运动。该方式将药物运动映射在x、y和z轴上以及结肠施加的倾斜角度。该药物可以评估肠道内内容物的运动方向(顺时针和逆时针)、速度和距离,从而计算结肠传输时间。最近的研究也证明了结肠运动模式与HRM研究结果一致。此外,还引入了MRI,因为它能够测量结肠游离水含量和结肠内容物的“流动性”。最近的动物研究表明,结肠能够适应内容物的各种物理特征,并根据或多或少的液体内容物的存在产生不同的运动反应。这些生理变量很可能在药物的溶解和/或吸收中发挥关键作用,这些药物在人类胃肠道的结肠部位释放。
胃肠道是一个复杂的器官,其环境随着时间的变化而变化。然而,对每个环节的难点和机会进行深入的理解对于实现最佳药物吸收和生物利用度(BA)是必要的(图2)。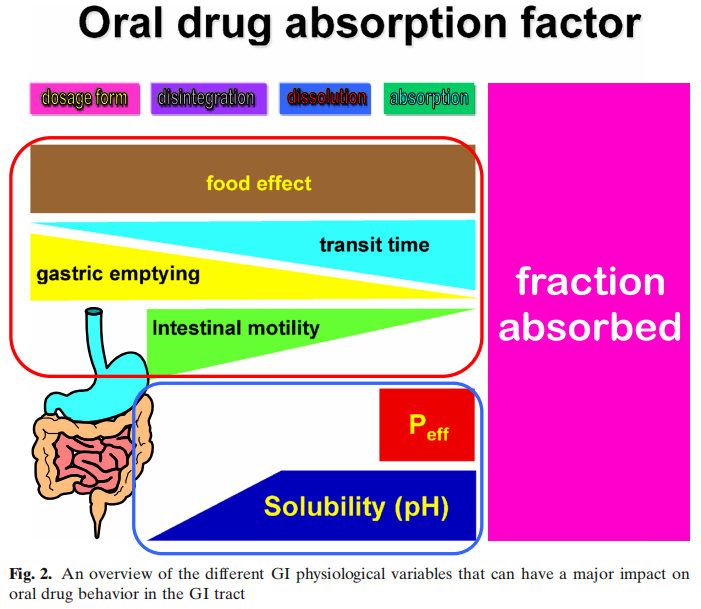
图2显示了影响吸收的多个因素,而没有考虑代谢或药物稳定性的影响。以蓝色方框表示的生物制药分类系统(BCS)侧重于渗透性和溶解性。然而,药物的溶解性和溶解度取决于红框中总结的生理因素。众所周知,运动效应和胃排空会对药物的性能产生影响,但在药物开发中很少考虑它们。相反,在过去的几十年里,人们对食物效应、pH效应和胆汁盐的增溶作用进行了深入的研究。然而,今天,对于一种通用的溶出介质仍然没有达成共识,这种介质可以用于药物开发和体外性能测试,以获得这些结果。早期研究评估了格列本脲(一种BCS II类药物)在生物相关介质中的溶解度。研究表明,当应用计算机模拟时,在胆盐介质(含有牛磺胆酸钠和卵磷脂)中增加的溶解度适合于建立体内-体外相关性(IVIVIVC)。在GastroPlusTM中建立了线性回归。参比制剂的回归系数为0.94。当使用生理性介质的溶出度值输入GastroPlusTM时,预测血浆Cmax和AUC的误差(%)分别为7%和14%。在水性介质(pH 6.5)中获得的溶解度相对于血浆Cmax和AUC的预测误差分别为38%和63%。后来,在生理相关介质(即FaSSIF)中开发了动态溶出方案,该方案再次显示出建立IVIVC的预测能力。然后将此动态流通装置溶出方案应用于孟鲁司特钠。同样,生理性介质与临床观察数据的拟合最好。这些早期研究在不考虑其他胃肠道因素的情况下成功地建立了IVIVC。在Almukainzi等人的一项研究中,研究了胃动力对美洛昔康药代动力学(PK)的影响。观察到两种制剂(常规制剂与快速溶出制剂)在啮齿类动物模型中给药时具有相似的PK结果。然而,当胃动力受损时,胃控制了药物的释放,因此,常规剂量的药物吸收是不同的。但是快速溶解制剂的PK接近健康状态下的结果。本研究表明,处方组成的差异在健康状态下不影响,但在疾病状态下可能导致显著差异。这项研究表明,在疾病条件下,胃能够对PK参数(如血浆Cmax和Tmax)产生负面影响,此外,人们普遍认为,对于许多药物来说,胃排空会影响空腹与餐后的PK。然而,很少有人关注GI运动对Cmax和Tmax的影响,这取决于给药时间和MMC阶段。这可能是由于用于量化和描述药物的PK行为的PK模型缩小了平均PK中单个的变异性。然而,如果同时监测肠蠕动和PK,则观察到的血药水平与肠蠕动之间的关系越来越显著。另一个影响药物吸收的因素是肠液的成分。胃肠道中的缓冲系统是以碳酸盐为基础的。在常规的药品质量控制和开发中,磷酸盐缓冲液起主要作用,而碳酸盐缓冲液很少使用。选择磷酸盐而不是碳酸氢盐似乎会影响肠溶制剂的体内性能。早期的报道显示肠溶产品在体内失败(1964年),直到今天的几十年里,体内研究才证实了这一点。此外,有证据表明,磷酸盐和碳酸盐缓冲液与肠溶包衣的相互作用似乎不同。很明显,重新评估已建立的体外测试方法对于获得体内相关性能以避免产品失败非常重要。除了缓冲液的性质外,下一个重要的区别是体外溶出方案中使用的缓冲液浓度与胃肠道中目前的缓冲液浓度以及药物溶出对肠道吸收的影响。多年来,两相溶出作为评估药物制剂体内性能的替代品。基于药物在有机层中的的渗透量,可以进行与所吸收相关的预测。然而,它对IVIVC的影响尚未得到充分认识。在最近的一项研究中,我们研究了布洛芬在药典以及与GI浓度相当的磷酸盐缓冲液的溶出行为。结果表明,布洛芬在药典条件下在不到15分钟的时间内完全溶解。然而,在低缓冲液浓度下,这一过程需要更长的时间,并且由于布洛芬的羧酸性质,介质的pH值发生了显著变化。然而,如果进行两相溶出试验,pH值随着时间的推移恢复到初始值。这再次证明了适应生理的体外测试对于获得体内状态是多么重要。只有这样才能确保体外方法能够预测体内性能。未来需要对这些方法转化为QC方法进行更详细的研究。最后一个方面涉及药物在体内的性能与体外行为无关。右美沙芬就是一个罕见的例子。这种药物在2小时内被吸收到80%以上,但需要大约15-20小时才能观察到Cmax。经典IVIVC将吸收与溶出相关联。然而,在这种具体情况下,IVIVC会产生误导。药物在肠道中快速溶解,并在15分钟内完全溶解。如前所述,80%的药物在2小时内被肠细胞吸收。药物在进入肠细胞后会被溶酶体吸收。作为弱碱,它在细胞质的生理pH值下具有高度亲脂性。由于药物会从肠细胞的顶端迁移到基底外侧,它可以穿过溶酶体的膜,进入ph值为微酸性的水环境。在这个细胞器中,弱碱变得更亲水,因此会被溶酶体包围。这就是为什么它在血液中出现的时间比它被吸收的时间要长。应该指出的是,对于这些特定的药物化合物,溶出度试验不能表征体内性能,因为药物产品的溶出度不能与血药水平直接相关。是生物系统及其特定环境和细胞室之间的药物分配决定了药物在中央室的浓度,而不是药物的溶解。总之,胃肠道药物吸收受到不同生理因素的高度影响。体外性能测试应考虑并包括适应生理环境的测试方法,以确定潜在的临床相关的剂型因素。BCS分类系统包括酸、碱和中性分子,可以帮助识别这些不同口服药物吸收的潜在障碍。为了满足所有这些标准,一个潜在的体外装置,可以模拟不同的胃肠道条件,如图3所示。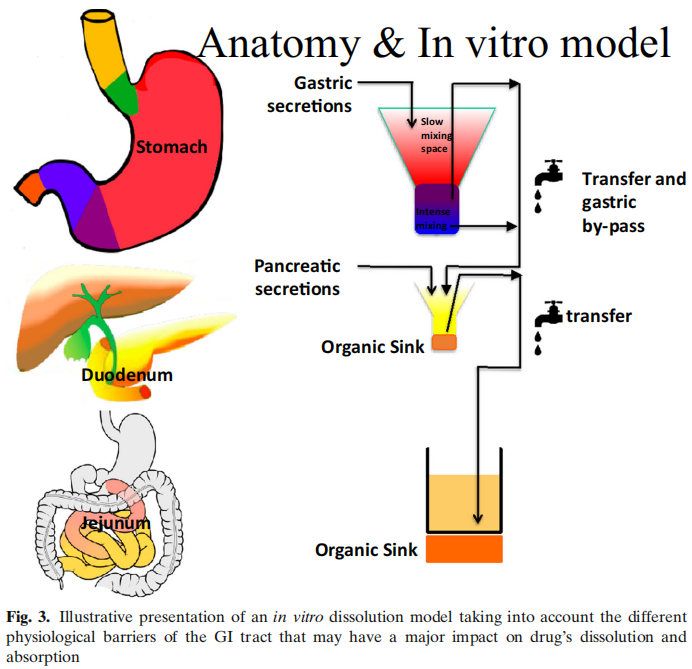
当两种药物不属于同一BCS分类时,即当它们吸收的限制因素不同时,固定剂量药物组合(FDC)产品的开发可能具有挑战性。在介绍的第一部分中,在不同分类体系的框架内讨论了探索组合产品中每种药物的生物药剂学特性的相关性。BCS标准框架,其中主要关注的是确定体外方法的非生物等效性(BE不等效)风险,这有助于处方的选择。为了理解药物的生物药剂学的限制因素,在可开发性分类系统(DCS)需要去修饰它的渗透性与溶解性及BCS。DCS考虑了小肠中较高的可用液体体积(500 mL)和人体肠液中的溶解度及溶解性分类。500mL的体积是基于共同给药时的送药液体体积以及胃肠道中液体体积计算的。另一个相关的补充是区分溶解度受限和溶出度受限药物,因为配方方法可能不同。可以根据药物的物理化学特性选择溶出试验来探索体内不等效的风险。为此,Tsume等人提出了BCS的分类系统。BCS II类药物分为中性(BCS IIc)、弱酸(BCSⅡa)和弱碱(BCSⅢb)。根据这些细分,用于预测体内行为的溶出试验不同于I类和III类,其简单的溶出仪(如USP II)就足够了,而II类和IV类则应包括隔室和吸收池,以增加体内的可预测性。为了适应这种需要,文献中提出了几种溶出体系,并描述了几种两相或多相的可切换的溶出体系。
在第二部分,讨论了制剂赋形剂的潜在影响,以及研究这些影响的实验临床前模型。在胃排空或肠道上,赋形剂可以影响膜通透性、代谢和胃肠道运动。在表I中,总结了一些具有有用参考文献的实验方法。
例如,十二烷基硫酸钠(SLS)对非索非那定肠通透性的影响通过Doluisio的闭环灌注法进行了表征,并在大鼠体内BA研究中得到了进一步证实,而在大鼠胃排空试验中,通过硫酸钡评估了由于赋形剂引起的胃排空变化与生物等效性(BE)研究失败的相关性。最后,使用BCS作为BE问题的风险评估工具的概念是在FDC开发的案例研究的帮助下提出的。缬沙坦/氢氯噻嗪仿制药在BE测试中两次失败,其中一种药物失败,另一种药物成功。使用胃肠道模拟器(GIS)进行的生物预测溶出试验的应用成功地再现了体内结果,因为由于制剂中山梨醇和SLS的不同水平,胃室内崩解的差异和肠道中溶出速率的差异是体内失败的显著原因。总之,FDC中药物的BCS和/或DCS分类是定义吸收影响因素和影响BA的相关生理变量的工具。对于含有不同BCS类别药物的FDC,有必要结合体外溶出方法和临床前模型来评估制剂性能。
FDC产品以固定剂量比将两种或两种以上活性药物成分(API)组合成药物剂型。FDC产品是根据组合规则批准的,该规则规定每个成分都应有助于产品的有效性,并且该组合在特定患者群体中也应是安全的。安全性和有效性数据可以是全部(新药申请)或部分(505(b))原始数据,也可以基于以前的报告(简称新药申请)。FDC产品比单一实体产品(SEP)的联合给药具有几个优势,如最大的患者依从性、提高的安全性和有效性、最大限度地减少滥用潜力以及降低患者成本。它们还为制造商提供了在产品生命周期中扩展知识产权和排他性的机会。另一方面,配制FDC产品带来了一些与原料药之间的不相容性以及与某些赋形剂的不相容相互作用相关的挑战。一些药物可能在另一种药物(盐酸胺碘酮-青蒿琥酯)存在下降解,另一些药物可能具有药物不相容性(辛伐他汀-替米沙坦),一些药物可能表现出非常不同的粘弹性特性(二甲双胍-格列本脲),以及其他可能在吸收(例如,肠道转运蛋白)或吸收后(例如,代谢酶、肾转运蛋白)水平上的相互作用。
世界卫生组织根据产品注册的监管要求,将FDC产品分为四种不同的情况:
①新的FDC产品具有与现有FDC产品相同的活性成分和剂量。
②新的FDC产品与单一实体产品(SEP)的具有相同的活性成分和剂量。
③新的FDC产品将具有既定安全性和有效性数据的原料药组合在一起,但尚未用于该特定适应症。新FDC产品包括具有既定安全和有效性的原料药的组合,但将用于不同的给药方案。
④新FDC产品含有一种或多种新的化学实体(NCE)。
为了将参比制剂(RLD)的关键临床数据与属于上述①和②的FDC产品的安全性和有效性联系起来,需要进行BE研究。虽然①的BE研究设计是标准方法,但在②中,将FDC产品的体内性能(例如,PK终点)与sep的联合给药进行比较。在这两种情况下,成功的BE表明API之间不存在(或类似)PK相互作用。然而,FDC产品的BE研究具有挑战性,因为(i)组合产品中PK受试者内在的变异性;(ii)不同浓度的非线性PK;(iii)药物制剂的相互作用;和(iv)当作为组合产品服用时,食物对活性成分PK的不同影响。这些考虑使生物豁免成为制造商满足BE要求的极具吸引力的机会。目前,WHO、FDA、欧洲药品管理局(EMA)、国际协调会议(ICH)和加拿大卫生部允许对仅含高溶解度API的立即释放(IR)FDC产品使用基于BCS的生物豁免。因此,含有BCS I类和/或III类活性成分的FDC产品可以申请生物豁免。
一般来说,溶解和组成要求与SEP的要求相同,但各司法管辖区之间存在一些差异。对于BCS I类API 的产品,FDA要求使用目前FDA批准的IR产品中存在的辅料,而EMA鼓励使用与参比制剂相似量的相同辅料。2018年ICH关于基于BCS的生物豁免指南指出,关键赋形剂(如聚山梨醇酯80、山梨醇)必须在参考产品的±10%范围内。另一方面,各司法管辖区对辅料可能对BCS III类药物产生的影响达成共识,例如各机构要求辅料在种类上(Q1)与参比相同,在数量上(Q2)与参比非常相似。FDA和ICH指南包含赋形剂(按功能)相对于参比制剂的成分允许差异表。如果产品是药物等效物,并且满足溶解和成分要求,则对①中FDC产品实施基于BCS的生物豁免是简单的。如果产品满足溶解和组成要求,则对①中FDC产品实施基于BCS的生物豁免是容易实现的。此外,如果有适当的理由,美国食品药品监督管理局可能会接受基于BCS的仿制药生物豁免。另一方面,基于BCS的②中FDC产品生物豁免给制造商和监管机构带来了一些挑战。首先,不同的单一实体RLD产品可能在不同的地区注册,这意味着制造商必须进行多次生物豁免研究才能在不同的司法管辖区获得批准。与FDA不同,EMA没有公布不同欧洲国家的RLD清单,这可能会进一步复杂化。其次,含有不相容原料药的FDC需要结合分离技术(例如,双层片、片中片等),以获得稳定的产品。在这种情况下,可能很难解释FDC产品和各自SEP之间的成分要求。第三,研究活性成分剂量差异较大(即剂量比>50%)的FDC产品的溶出方法在分析上可能具有挑战性。当API在生理范围内表现出不同的pH依赖性稳定性的情况下,这可能会更加复杂。此外,RLD SEP可能使用不同的溶出装置(如篮或桨),因此制造商可能必须为一种FDC产品开发和验证两种溶出方法。第四,FDC产品中可能存在药物-药物或药物-辅料PK相互作用(DFI)的因素,当SEP共同给药时,这些相互作用可能不同或不存在。美国食品药品监督管理局和欧洲药品管理局都发布了关于在转运蛋白水平上研究药物相互作用(DDI)的指南。美国食品药品监督管理局还发布了研究体外转运体介导的DDI的方法学建议。虽然各机构要求申办者研究肠外排转运体介导的DDI(即p -糖蛋白、乳腺癌耐药蛋白),但目前尚无关于研究肠摄取转运体介导的潜在DDI的公开建议。这似乎令人惊讶,因为众所周知,肠道表达的摄取转运蛋白与大量结构不同的化学和治疗类药物有相互作用。此外,越来越多的证据表明,药物辅料可以在体外和原位抑制肠道转运蛋白的外排和摄取。包括聚乙二醇类表面活性剂,司盘和聚乙二醇。
虽然人们一致认为DDI或DFI可能与BCS I类药物的临床相关性较小,但也有人担心这些相互作用可能会极大地影响低渗透性药物的口服吸收。FDC产品还为开发一系列可用于通过剂量滴定优化治疗的给药方式提供了机会。中等规格和低规格可适用于基于剂量规格(DS)的生物豁免,前提是至少有一种规格(通常是最高的)在体内成功证明了与参比制剂的BE等效。基于剂量规格的生物豁免适用于不符合基于BCS的生物豁免条件的原料药和IR以外的药物形式(即改良释放、延迟释放)。各司法管辖区对基于DS的生物豁免的共同要求是治疗剂量范围内的线性PK,有机会在最高和最低规格之间采用括号法,并且制造过程相同。FDC的剂量范围将取决于研究药物的相加或协同作用。药物之间的相互作用在药物-药物相互作用和PK-PD研究中进行评估。随后,暴露反应模型可用于2B期剂量选择。与基于BCS的生物豁免一样,基于DS的生物豁免要求是SEP的扩展。表II和表III总结了基于DS的生物豁免的FDA和EMA的要求和溶出方法建议。表II和表III中的数据表明,在美国和欧洲市场寻求基于DS的生物豁免的制造商在满足基于分离技术的FDC产品(如双层片剂)的成分要求方面可能面临挑战,因为EMA将每一层视为一个单独的实体,而FDA将双层片剂视为单个单元。此外,在单一单位的情况下,原料药之间剂量差异较大的FDC产品可能很难满足美国食品药品监督管理局和欧洲药品管理局的比例要求。更具体地说,EMA规定,为了计算API/辅料的比例,必须将其他API视为辅料。然而,尚不清楚是否必须将其他API视为辅料。同样,对于如何在双层片中考虑其他API,FDA也没有具体的建议。这些差异可能会阻碍FDC产品在美国和欧洲同时注册申报。此外,虽然美国食品药品监督管理局要求对不同规格中的最高剂量进行BE研究,但欧洲药品管理局要求除最高规格外,还需要对最低规格进行研究。最后,不同司法管辖区之间存在不同的参比制剂,增加了申办方在不同地区寻求批准时需要的不同的研究数量。
Amitava Mitra 博士(Sandoz,Inc.,A Novartis Division)讨论了在实现含有两种或两种以上活性成分的FDC产品的BE过程中克服挑战的关键点和策略。这些产品的活性成分可以通过不同的药理学途径发挥作用,并具有增加/协同作用、减少每种活性成分的剂量和提高患者依从性的优势。帕金森氏症药物左旋多巴(Levodopa)的新型fdc是利用新技术和新机制改善旧药物临床疗效的一个例子。
然而,将多种活性成分组合可能会使其单独的生制药和PK行为复杂化。由于药物释放的不同,控制或修饰释放FDC产品的开发确实增加了额外的挑战。释放曲线的变化可能会改变API的生物药剂学和药代动力学。还讨论了严格审查FDC中单个药物的物理化学和生物药剂学特性及其对PK的影响的重要性。全面了解单个药物的PK特性以及FDC中的配方变量是同样重要的需要考虑的因素。与FDC战略相关的BA研究非常重要,值得鼓励。然而,有太多变量的且不充分研究可能会进一步混淆已经很复杂的问题,应该避免。在设计BE研究时,应充分考虑来自所有化合物的所有物理化学、生物药剂学和PK数据。可以考虑针对高变异药物的BE研究设计,如收紧BE或交叉重复设计。利用从不同但协同的技术中获得的知识,如体外溶解度/溶解性研究,吸收模型和IVIVC,体内临床前动物模型,以及可用的体内临床数据,对于给定组合的FDC策略的成功至关重要。讨论了两个案例研究,其中使用口服吸收建模、溶出度数据和临床PK数据成功开发FDC产品。在第一个案例研究中,讨论了三联产品的开发,其中一种活性成分具有高度可变的Cmax,而另一种活性成份由于胆汁分泌和吸收缓慢而具有较长的Tmax。在这种情况下,口服吸收建模是理解配方变化对三种活性物质PK的影响以及最终对FDC产品开发的影响的关键点。在第二个案例研究中,讨论了双组份产品的开发,其中一种活性成分是弱碱,具有高的受试者体内CV和陡峭的pH溶解度。在这种情况下,来自几项相关BA研究的数据以及对PK和生物药剂学特性的深入了解有助于FDC的成功开发。
尽管其中一个目的是以固定的剂量比组合药物,以简化慢性病的治疗并提高患者的依从性,但人们普遍认为,这一原理不能成为任何开发或配方设计背后的唯一目标。对世界各地司法管辖区FDC产品备案法规的概述表明,这方面的进展相当缓慢。总的来说,通过结合先前批准的单产品或从NCEs的共同配方开始开发FDC产品可以遵循有限的监管途径。根据美国法规,FDC的监管基本原则在《联邦法规》和指南中进行了描述,概述了FDC产品批准的要求。2013年引入的开发指南从监管角度反映了这些药品的重要性。该指南描述,如果新FDC的剂量、拟定的治疗适应症、或者所需要的临床数据没有变化,则药物疗效可以依赖于BE测试。FDC产品可以遵循以下监管途径之一:505 b(1)、505 b(2)或505 j,涵盖从新药开发到仿制药开发的所有可能性。另一方面,EMA发布了关于FDC产品临床开发的若干指南,指明了任何FDC开发的拟治疗用途和适应症。该指南描述了三种可能的情况,并对疗效证明提出了具体要求:(1)如果对拟组合中包含的一种或多种药物存在不良反应,则使用FDC产品作为附加治疗。如果联合用药对潜在的临床后果构成威胁,则可能需要进行药物-药物(DDI)或PK的相互作用研究,(2)当寻求减少药片数量负担时,用FDC产品替代。如果FDC产品以不同的时间间隔给药,则需要进行BE测试,并应特别注意;如果FDC以前没有用于任何特定的适应症,开始FDC治疗,则临床和pk试验以及DDI研究都应在批准前进行并提交。在拉丁美洲,自2010年以来,FDC产品的注册只有一个具体指南,它描述了FDC产品的定义、申报的一般考虑以及取决于拟定剂量方案或拟联合药物的监管要求。在以下条件下可以获得FDC批准:(1)FDC产品含有与同时使用的单一产品相同的活性物质、剂量和给药方案;因此,安全性和有效性是众所周知的;为了证明疗效,BE研究可能就足够了;(2)与“(1)”中的条件相同,但FDC产品将用于新剂量或新的治疗适应症,因此需要进行III期临床试验;(3)该组合含有一种或多种新的活性成分,需要进行I、II和III期临床试验才能获得批准。一般来说,对于FDC产品的注册没有全球适用的指南,但针对特定的治疗类别和世卫组织技术报告中描述的四种一般情况,旨在指导制药公司在欠发达司法管辖区开发、批准和销售FDC产品。尽管在大多数司法管辖区,仿制药和复方药提交途径似乎已经足够多,但是新的复方制剂仍然需要临床前与临床数据即使FDC药物中单个的药物是已知的或者是新发现的。然而,监管者仍然坚持认为,世界各地不同的司法管辖区应更加重视便利性/合规性,将其作为开发FDC产品的基本原理,要么包含授权/新药实体,要么只考虑患者满意度或降低/控制医疗成本。如果允许仿制药开发,FDC产品的BE研究设计应考虑与单独给药相同的原则,寻找每种FDC活性成分及其各自的参比FDC或参比单品的PK的等效性。在一点上,这是十分重要的意识到PK相互作用可能会有更加关键的结果在FDC产品上相比于同时给予相同药物的单一产品。总之,在比较获得FDC产品批准的司法管辖区时,似乎有必要在FDC产品过于谨慎的注册、有效性证明与FDC产品可能带来的巨大公共卫生效益之间取得平衡。在FDC产品开发、注册的现有技术指南、理解和应用方面实现全面的平衡仍然是一个未解的问题。
对于没有FDC开发经验的配方科学家,讨论了两个决策树,以选择最合适的配方开发策略。第一个决策树与FDC产品的配方设计有关(图4)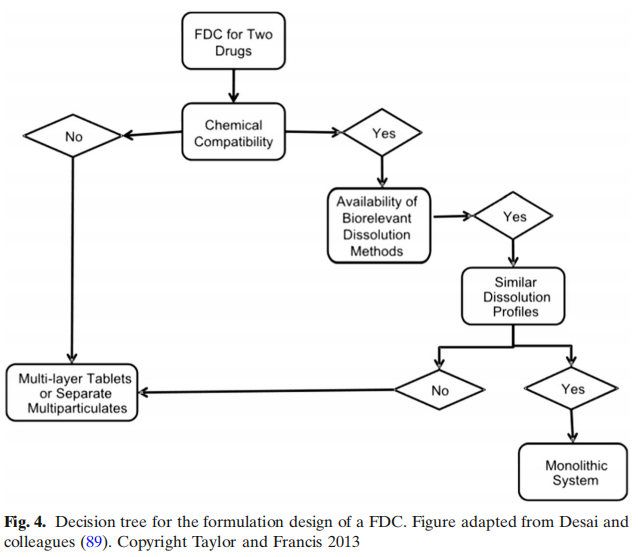
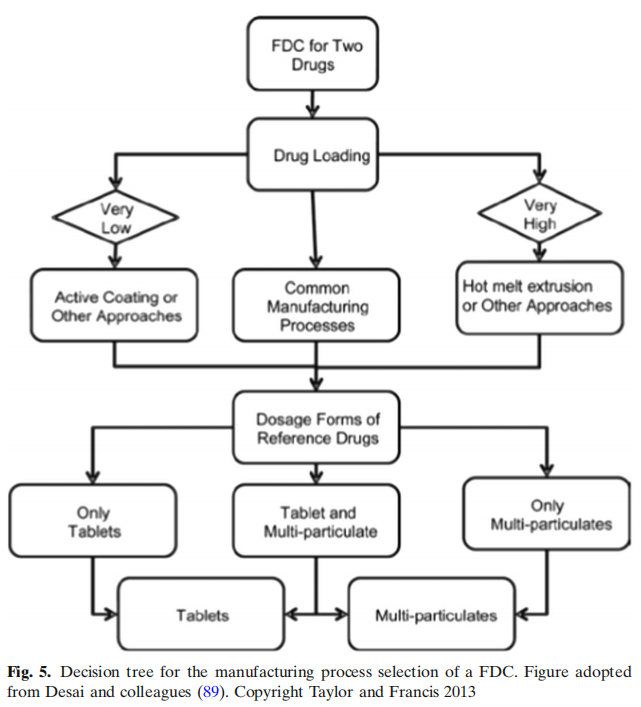
制剂中的药物载药量决定了生产工艺的选择。如果载药量大,可采用热熔挤出或双层法制备。对于低载药量的制剂,提出了一种活性成分上药包衣方法。制造工艺选择的关键因素之一是制药科学家先前的经验。单片制剂系统,即两种药物合并在一个单剂量单位中,被认为是最简单的制剂方法。然而,在一个案例研究中,第二种药物氢氯噻嗪(HCTZ)被添加到现有的高血压药物配方中。结果表明,聚维酮(一种粘结剂)和波洛沙姆(一种润湿剂)通过在有效水分中溶解HCTZ,在加速储存条件下引发HCTZ降解。用淀粉1500代替聚维酮,解决了稳定性问题,并去除了泊洛沙姆,对BE研究没有不利影响。对于双层片剂处方方法,通常用于将两种不相容的药物分开或保持两种药物释放曲线,很少有关键的处方因素被提出。这些因素包括选择具有高脆性的赋形剂,如第一层中的乳糖;第二层中的可塑性材料,如微晶纤维素,以及两层的重量比不超过1:6。还强调的是,第一层的压力应该能够在不牺牲表面粗糙度的情况下减小体积,而表面粗糙度对于第二层的粘附是必不可少的。提出了关于双层片处方方法的两个案例研究。在第一个案例研究中,通过添加1% w/w的二氧化硅,二甲双胍缓释制剂的可压缩性得到改善。在第二个案例研究中,两种不同等级的气相二氧化硅在双层片剂配方中的表现不同。Aerosil 200没有引起层分离,但Aeroperl 300引起了分层。由于其更大的表面积,在任何湿度水平下,Aeroperl都能吸附相对大量的水分,但当湿度降低时,它不能保留水分。相比之下,Aerosil吸附的水分相对较少,但由于其大孔径,它能保持水分。据推测,Aeroperl未保留的水分可与其他层赋形剂(如交联聚维酮)相互作用。提出的第三种配方技术是包衣技术。包衣层也可用于维持两种单独的释放曲线,并分隔两种不相容的药物。一个案例研究展示了酸碱敏感分子是如何稳定的,选择可以降低API与辅料密切接触包衣材料。例如,对于100mg片剂,将1mg药物与99mg赋形剂一起放置,1mg的药物可以与99mg的赋形剂反应;然而,如果将1mg的药物与9mg的包衣材料一起放置,则可用于反应的量急剧减少。这也是生产压缩敏感药物片剂的有用技术。虽然活性涂层是有用的,但它没有像其他技术那样广泛应用,因为它提出了两大挑战。第一个挑战是如何检测包衣终点,从而生产出具有拟定作用的片剂。如果包衣过程过早停止,片剂可能药效不足。另一方面,如果涂层停止得晚,片剂可能会超级有效。第二个挑战是含量均匀性(活性成分涂层均匀性)。含量均匀性可能受到各种工艺参数的影响,如包衣锅生产批量、包衣时间、喷枪的数量和包衣质量。提出了模型参数与工艺参数相联系的数学模型。结果表明,该模型能准确预测200 ~ 1450mg不同形状片剂在450kg商业规模下的包衣均匀性。总之,决策树对于探索FDC最合适配方和制造过程非常有用。FDC的每种配方方法都有其独特的挑战,但正如各种案例研究所示,有可能克服这些挑战,利用各种过程分析技术(PAT)开发坚固的配方和商业上可行的制造工艺。
组合药物在《联邦法规法典》21 CFR 3.2 (e)中定义为药物-药物联合产品的类别。这些产品可以是两种已批准药物或者两种已批准以上药物或者是在研药物与已批准药物或者两种在研药物或者两种以上在研药物组合在一起共同开发。最终产品可以是FDCs、共包装产品或分开的单个的药品再一起使用。开发这些产品的原因之一是针对同一疾病的药物(例如抗病毒和咳嗽/感冒药产品)的加性/协同效应。开发这些产品的原因之一是针对同一疾病的药物(例如抗病毒与咳嗽/感冒药物产品)的相加/协同作用。有时,当两种药物具有互补的作用机制时,它们会被作为FDC产品开发用于同一疾病。例如,将β内酰胺与β内酰胺酶抑制剂相结合,可以选择性杀死对β内酰胺具有耐药性的细菌。有一些FDC的例子,某成分减少其他成分(例如萘普生/埃索美拉唑缓释片)的不良事件。大多数FDC是口服的,但也有吸入性的例子(例如,用于慢性阻塞性肺病(COPD)的噻托溴铵/奥达特罗)与眼科产品(例如用于降低眼压的奈舒地尔/拉坦前列素)。本报告的目的是概述FDC开发中的临床药理学考虑。FDC的开发给药物开发人员带来了有趣的挑战。如果正在开发两种或两种以上的新分子实体(NEMs)作为FDC,则通常需要对药物给药剂量进行研究,以确定每种药物的合适的给药剂量。药物管理方面的挑战,如食物对FDC的影响,通常需要加以解决。当FDCs中提出的各种药物在进食和禁食条件下的给药要求不同,或者药物给药频率不同时,这种情况可能会变得更加复杂。这些情况通常需要更仔细地研究FDC的配方和进行额外BA研究。考虑到配方的不灵活性,特定人群中FDCs的剂量调整可能存在问题。作为FDC开发计的典型研究是关乎BA研究。FDC BA研究的目的是将FDC中每种活性药物成分或治疗部分的吸收速率和吸收程度与作为单独的单成分制剂同时给药的的吸收速率、吸收程度进行比较21 CFR 320.25(g)。通常,建议在复方产品最高规格并且与相匹配的单个药物的制剂下对比研究复方制剂与单活性成分药物的二周期、单剂量空腹研究。替代性研究设计,如比较联合用药产品与单一成分药物产品的单独给药的治疗研究设计,也可能是合适的。单剂量、食物影响这样在FDC产品上的研究用来评估食物对FDC的影响。本文介绍了与支持FDC批准的研究相关的BA研究案例,以及基于析因设计研究的FDC与单独给药的例子。FDA指南题为“联合开发两种或两种以上的新药”,列出了可能适合进行因子设计研究以确定FDC中单个成分的作用的情况。
FDC产品的市场准入具有挑战性,一方面是要实现单个药品的协同给药,另一方面也是由于配方方面的挑战(原料药的相容性、剂量)。然而,我们不应忽视胃肠道生理学对口服药物行为的影响,这可能导致受试者间差异,从而可能导致BE研究的失败。因此,在监管当局的指导方针的支持下,确定制剂开发战略与临床评价之间的关系是很重要的。此外,体外溶出测试的贡献可以在某种意义上帮助监管机构对FDC产品批准的决策,因为这些模型可以模拟在胃肠道吸收过程中起关键作用的潜在的GI变量。从学术角度来看,当制药公司分享他们的非BE配方(即临床失败)时,可以对这些临床相关的溶出模型进行优化和验证。当他们这样做时,潜在的问题可能被解开,配方科学家在制定FDC产品时应考虑到这些问题。






