

时间:

翻译:工业药剂发烧友Summer
审核:华溶应用中心
长效注射混悬剂难溶性药物晶体在体内缓慢溶解,可将药物体内释放时间延长到数周至数月。迄今为止,FDA已经批准了大约10种长效注射混悬剂,但大多数都没有等效的仿制药,很可能是由于复杂的处方工艺以及建立体外-体内相关性(IVIVC)方面的困难。基于动物模型的A级IVIVC已被证明可用于具有多相释放特征的复杂长效微球。长效注射混悬剂的释放特性相对简单,只有一个药物溶出阶段,因此可建立IVIVC。为了建立长效注射混悬剂的体内外相关性,以长效注射醋酸甲羟孕酮(Depo-SubQ Provera 104)为上市参比制剂,制备了四种活性成分相同但处方和工艺(药物粒度和辅料来源)不同的醋酸甲羟孕酮长效注射混悬剂。然后使用两种基于USP装置二(带有浸没池)和USP装置四(带有半固体适配器)改进的体外释放测试方法,通过兔模型研究体内释放,并使用USP装置四法获得了体外释放曲线,成功建立了A级IVIVC。
长效注射混悬剂难溶性药物晶体在体内缓慢溶解,可将药物体内释放时间延长到数周至数月。迄今为止,FDA已经批准了大约10种长效注射混悬剂,但大多数都没有等效的仿制药,很可能是由于复杂的处方工艺以及建立体外-体内相关性(IVIVC)方面的困难。基于动物模型的A级IVIVC已被证明可用于具有多相释放特征的复杂长效微球。长效注射混悬剂的释放特性相对简单,只有一个药物溶出阶段,因此可建立IVIVC。为了建立长效注射混悬剂的体内外相关性,以长效注射醋酸甲羟孕酮(Depo-SubQ Provera 104)为上市参比制剂,制备了四种活性成分相同但处方和工艺(药物粒度和辅料来源)不同的醋酸甲羟孕酮长效注射混悬剂。然后使用两种基于USP装置二(带有浸没池)和USP装置四(带有半固体适配器)改进的体外释放测试方法,通过兔模型研究体内释放,并使用USP装置四法获得了体外释放曲线,成功建立了A级IVIVC。
IVIVC是一种预测数学模型,用来研究不同剂型药物的体外溶出与体内释放之间的关系。成功的IVIVC可用于制定释放/溶出测试规范,当建立了人体内的A级相关性时,可代替体内BE研究。在药物初次批准过程中以及在获批后出现微小变更(例如生产场所变更)时,IVIVC可以减轻BE研究的临床负担。迄今为止,关于IVIVC的建立、评估和应用,仅有一项针对缓释口服剂型的FDA指南。因此在过去20余年里,科学家们不得不依赖这一指南来开发非口服制剂。一项针对IVIVC研究的行业调研显示,半数受访者很少或从未建立过非口服剂型的IVIVC模型。FDA已批准的长效注射混悬剂产品中,只有Invega Sustenna提供了针对不同粒径的临床A级IVIVC 。近年来,由于人体研究相对困难并且昂贵,非临床动物模型已被用于非口服剂型IVIVC的研究,可能为人类IVIVC的建立铺平道路,例如各种聚乳酸-甘氨酸微球制剂如地塞米松、利培酮、纳曲酮、醋酸亮丙瑞林,已经通过兔模型成功建立了A级(点对点)IVIVC。
在IVIVC模型研究中,根据所选制剂的药代动力学特征,动物模型或人体受试者的体内吸收(血药浓度-时间变化)呈现出恒定的规律。但当使用不同的体外释放/溶出方法(设备、释放介质、温度、搅拌速度和上样方式)时,所选制剂的体外释放曲线可能会有所不同。因此,选择合适的体外释放/溶出方法来建立IVIVC至关重要。尽管FDA已经推荐了一些长效注射混悬剂的体外释放检测方法,但它们的持续时间较短(30分钟至2天),可能不足以建立用于长效注射混悬制剂的IVIVC(体内有效持续时间为数周至数月)。本文开发了一种能够延长注射混悬剂体外释放时间的方法,有望用于IVIVC的研究。
在IVIVC模型研究中,根据所选制剂的药代动力学特征,动物模型或人体受试者的体内吸收(血药浓度-时间变化)呈现出恒定的规律。但当使用不同的体外释放/溶出方法(设备、释放介质、温度、搅拌速度和上样方式)时,所选制剂的体外释放曲线可能会有所不同。因此,选择合适的体外释放/溶出方法来建立IVIVC至关重要。尽管FDA已经推荐了一些长效注射混悬剂的体外释放检测方法,但它们的持续时间较短(30分钟至2天),可能不足以建立用于长效注射混悬制剂的IVIVC(体内有效持续时间为数周至数月)。本文开发了一种能够延长注射混悬剂体外释放时间的方法,有望用于IVIVC的研究。
2.1 材料
DMSO (USP级)购自Sigma-Aldrich(St.Louis,MO,USA)。戊烷,磷酸氢二钾,含0.1%甲酸的乙腈(v/v),LC/MS级Optima™和含0.1%甲酸(v/v)的水, LC/MS级Optima™购自Fisher Scientific(Hampton,NH,USA)。Depo SubQ Provera 104mg醋酸甲羟孕酮注射混悬液(104mg/0.65mL)购自Pfizer Inc。醋酸甲羟孕酮(微粉化,USP级)购自Spectrum Chemical Manufacturing Corp(New Brunswick,NJ,USA)。甲羟孕酮17-乙酸酯-d3(MPA-d3)购自TLC Pharmaceutical Standard ltd(Ontario,Canada)。MiniCollect 0.8ml肝素分离管购自Greiner Bio-One North America Inc。(Monroe,North Carolina,USA)。除非另有说明,否则所有材料均为分析级。
2.2 MPA混悬液的制备及体外释放数据
首先用四种方法制备MPA混悬剂 :使用MPA原料药制备的制剂1(F1);使用重结晶制备的制剂2(F2)(反溶剂法:丙酮与水之比为1:1,分别作为良溶剂和反溶剂);通过探针超声处理制备的制剂3(F3);制剂4(F4)除了使用不同来源的PEG 3350(F1:来自Spectrum Chemicals;F4:来自BASF)外与F1相同。使用马尔文粒度仪测定所有制剂的粒径,记录Dv10,Dv50和Dv90,所有实验重复三次,然后计算跨度值((Dv90-Dv10)/Dv50)来评估混悬剂的粒度分布,所有数据均以均数±标准差表示。
所有MPA混悬剂的体外释放数据均来自之前的一份报告,其中使用了两种装置,分别为带有浸没池(4cm2接触面积)的USP装置二(推荐使用华溶仪器DS-1206AT全自动取样溶出系统)和带有半固体适配器(1mm深度)的USP装置四 (图1)(推荐使用华溶仪器DS-7CP流池法溶出系统)。
2.3 MPA混悬剂的体内释放研究
2.3.1 MPA混悬剂的动物研究
以兔为模型研究了MPA混悬剂和商业产品Depo SubQ Provera 104在体内的释放情况。将体重约4kg的雌性新西兰白兔随机分配到每个制剂组(n=6)。将每种制剂以26mg/kg的剂量皮下注射(0.65mL)。为了获得药物PK参数,MPA药物溶液(在DMSO中)也以4 mg/kg的剂量进行静脉注射(n=6)。以预定的间隔从耳缘静脉收集血液样品并置于肝素分离管中。样品在5000rpm的速度下离心5分钟得到血浆,然后在-80℃环境下储存直到分析。本研究动物模型的选择是基于以下几点原因:(1)PK研究的持续时间长达3个月,为确保有足够的血容量用于长时间的连续采样,认为较大的动物(如兔子)比较合适;(2)MPA用于避孕,因此选择雌性动物。为保证动物试验的安全性,本研究所采用的给药剂量均按相关文献规定,并确保给药后的血药浓度能被检测出来,本研究按照康涅狄格大学机构动物护理和使用委员会(IACUC)审查和批准的方案进行。
2.3.2 样品制备
超高效液相色谱(UPLC)
采用Vanquish UPLC系统(Thermo Fisher Scientific, Waltham, MA, USA)进行样品分离。并使用Kinetex EVO C18 (50 mm*2.1 mm, 2.6µm)和带有SecurityGuard™超滤筒的预柱(Phenomenex Inc, Torrance, CA, USA),将柱温箱温度设置为30℃。流动相由两种溶剂混合物组成,A为含0.1% v/v甲酸的水;B为含0.1% v/v甲酸的乙腈; LC/MS级Optima),用8分钟梯度法(表1)以0.3 mL/min的流速洗脱系统,进样体积为20μL。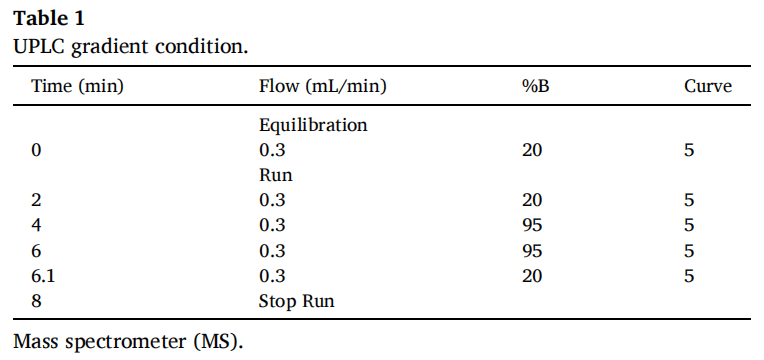
在全扫描模式下使用配备OptaMax™NG电喷雾(H-ESI)离子源(Thermo Fisher Scientific, Waltham, MA, USA)的Thermo Scientific™高分辨率Orbitrap Exploris™480质谱仪 (质量范围:2500 - 425.00m/z,分辨率:120,000)。对MS参数进行优化来获得目标药物分子的最佳强度。离子源参数设置如下:喷雾电压为3700V的正离子模式;护套气体流量为70Arb;辅助气体流量为23Arb;扫掠气体流量为5Arb;离子转移管温度为400°C;汽化器温度为525°C。方法总时间为8min。药物MPA和内标(MPA-d3)的亲本离子质量分别为387.2525m/z和390.2700m/z。通过Thermo Scientific Xcalibur™软件(4.4版本)进行数据采集和分析。
MPA和内标的保留时间均为4.81min。除最低浓度(0.5ng/mL)的回收率约为70%外,其他浓度MPA的回收率均在90%-101%范围内。校准曲线建立在0.5-50 ng/mL和50-1000ng/mL范围内,线性良好(R2>0.9995),准确度小于15%,精密度小于10%。
2.4 统计分析
使用OriginPro 2017软件(OriginLab Corporation)进行线性回归和拟合。数据以平均值±标准偏差(SD)表示。
2.5 IVIVC建立
为了研究用于MPA混悬剂的IVIVC,使用Phoenix WinNonlin软件和IVIVC工具包(版本6.4,Certara Inc.,NJ,USA)进行数据分析。目前的研究集中在A级IVIVC(体外溶出度与体内释放速率之间的点对点关系),因此它是最具参考性的,以下是数据分析的三个主要步骤。
步骤1:体外数据输入
步骤2:体内释放数据输入
在软件中通过数据反卷积处理获得所选MPA混悬剂的体内释放曲线。以静脉注射剂的药代动力学特征为参考。设定单位脉冲响应(UIR)指数的最大值为2,采用Akaike模型进行数据反卷积。
步骤3:预测和验证相关性
通过软件计算得到吸收比例因子(scaling factor for absorption)、时间比例因子(scaling factor for time)、时移因子(time shifting factor),用不同的缩放和/或移位因子建立体外和体内释放数据之间的相关性,在此基础上对所建立的IVIVC进行验证,并对AUC和Cmax的预测误差百分比(%PE)进行分析。根据FDA口服缓释剂型IVIVC研究指南 ,IVIVC % PE的合格标准是:(1)对于体内预测性,Cmax和AUC的平均% PE≤10%。此外,每种制剂的%PE不应超过15%;(2)如果体内预测性不确定,则应通过体外预测性来最终确定IVIVC是否能够替代BE;(3)对于体外预测性,Cmax和AUC的%PE应≤10%。
3.1 粒径与粒径分布
表2列出了MPA混悬剂(F1、F2、F3和F4)及RLD的粒径和粒径分布。排列顺序如下:F3
3.2 所有制剂的体内药物释放
以家兔为实验对象,对MPA混悬剂和RLD进行体内药物释放研究。为了获得PK曲线,所有制剂均经皮下注射 (图2B)。还进行静脉注射药物溶液(图2A)用于反卷积处理获得PK参数。为了获得PK曲线的清晰视图,图3A显示了平均值曲线。使用Phoenix WinNonlin对所有制剂的体内释放曲线进行反卷积。反卷积建立在非区室分析(NCA)的基础上,详细的方程推导可以在(Certara.com)中找到。为便于比较,还展示了先前报道的测试制剂的平均体外释放曲线(图3CD),粒径是分散体系(混悬剂、纳米颗粒、脂质体)中的重要属性,本研究重点考察了药物粒径对长效注射混悬剂在体外和体内释放特性的影响。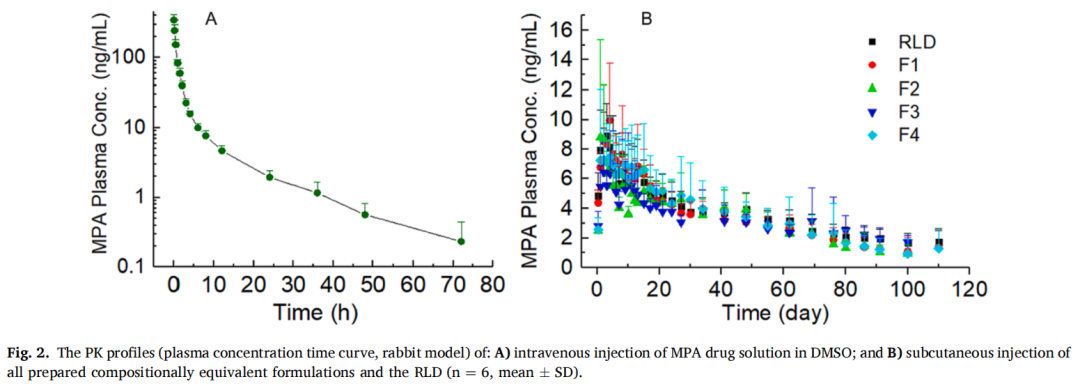
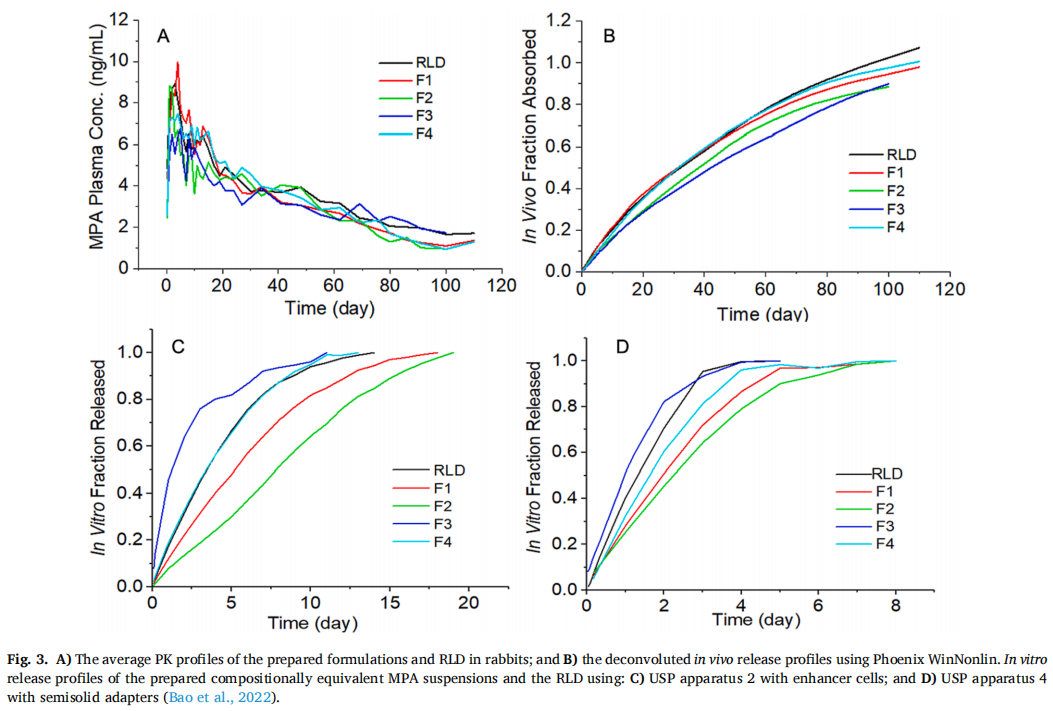
正如预期的那样,药物的粒径显著影响了体外和体内的释放曲线。粒径越大,在体内外的药物释放速率越慢。F1,F2,F4和RLD的体内与体外释放显示出相同的次序(图3CD)。但是F3并没有遵循相同的规律,它的粒径最小,预计在体内释放最快,相反,F3在体内药物释放最缓慢。进一步的研究表明,F3在体外并不稳定,室温下随着时间的推移容易发生聚集,其粒径(Dv50)在5天的时间内一直增加到20 μm左右(图4A)。此外,F3的跨度值也从2.61下降到1.5以下 (图4B),表明F3的粒径分布随着时间的推移而变窄。因此,F3颗粒在注射部位的聚集是造成体内释放缓慢的重要原因之一。此外,也有可能F3在体内快速溶解,然后在注射部位沉淀或再结晶,形成了更大粒径的颗粒,从而减缓药物释放。由于建立IVIVC的最基本原则是体外和体内的结果应一致,因此F3并未包括在体内外相关性的研究中。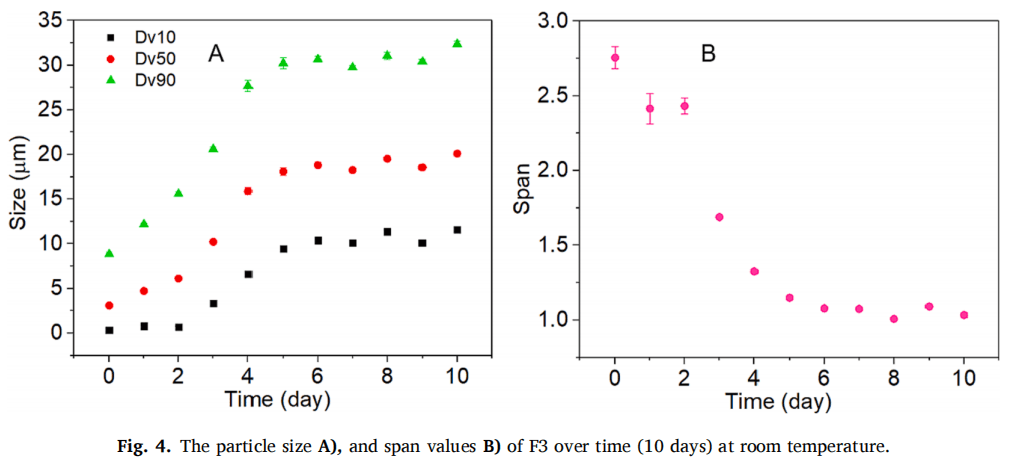
通过调整药物粒径(F1、F2和F3)和改变PEG 3350的赋形剂来源(F1和F4),制备了具有不同释放速率的MPA混悬剂,虽然使用完全相同的悬浮介质,但药物颗粒不一定具有物理稳定性,以探针超声法制备的F3为例,在常温常压条件下粒径随时间增长,初始粒径(Dv50)约为3µm,到第5天粒径约为20µm。在体外释放试验中并未观察到粒径的变化,这很可能是因为在漏槽条件下释放介质具有稀释效应,但在兔体内释放实验中观察到药物颗粒的聚集效应。这些结果说明了不同的工艺条件可以影响粒子的稳定性,所以在不同工艺条件下制备混悬剂是具有挑战性的,表明了精确控制长效注射混悬剂粒径的重要性。
3.3 MPA混悬剂的IVIVC研究
根据之前关于MPA混悬剂释放测试方法开发的报道,使用带浸没池的USP装置二(图3C)和带半固体适配器的USP装置四(图3D)显示出良好的释放特性,可以尝试建立A级IVIVC。使用两种装置可获得较长时间释放曲线 (至少一周),具有可接受的再现性和区分不同粒径MPA混悬剂的能力。在A级IVIVC的研究中,理想情况是体内释放部分与体外溶出部分之间有1:1的关系。如果体外释放持续时间接近制剂的体内疗效持续时间,则有可能建立无时间缩放和转换或最小时间缩放和转换的IVIVC。与目前FDA推荐的用于其他长效注射混悬剂产品的方法(30分钟至2天)相比,这两种方法体外释放持续时间长得多(USP装置二为2周,USP装置四为1周),但仍比体内疗效持续时间(3个月)短得多(图3CD)。
使用WinNonlin IVIVC工具包进行MPA混悬剂的IVIVC研究,将体外数据与所选制剂的体内释放数据进行比较。
3.3.1 使用USP装置二(带浸没池)获得的体外释放数据
首先,从带浸没池的USP装置二中获得了四种制剂F1, F2, F4和RLD的体外数据。F2、F4和RLD作为体内制剂,F1作为体外制剂,采用双Weibull模型拟合效果最佳。通过缩放和移位的方法,吸收分数和释放分数之间的IVIVC线性回归拟合系数(R2值)为0.9377(图5A)。使用建立的IVIVC将预测与观察到F1在体内的释放情况进行比较(图5B)。结果表明两条曲线在一些时间间隔内重叠,但在其他时段具有明显的区别。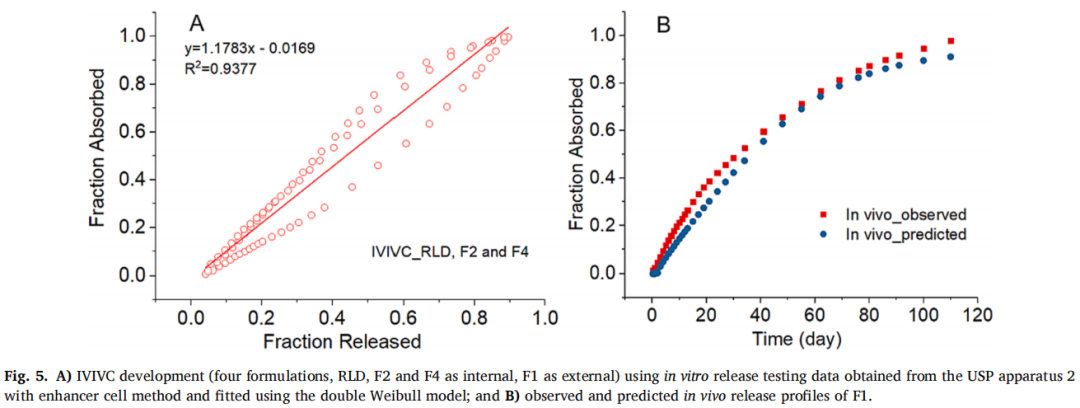
吸收比例因子(scaling factor for absorption)和时间比例因子(scaling factor for time)分别为0.91和0.17,时移因子(time shifting factor)为0.31。IVIVC验证总结如表3所示。对于体内预测性,AUC的%PE符合标准(个体%PE<15%,平均%PE<10%),但Cmax的%PE>10%。因此使用USP装置二(带浸没池)获得的体外释放数据并不能建立A级IVIVC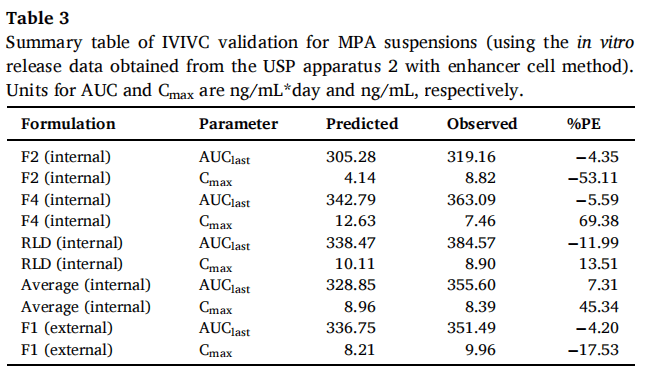
3.3.2 使用USP装置四(带半固态适配器)获得的体外释放数据
使用USP装置四方法获得体外释放数据,选择三种或四种制剂用于IVIVC的研究。
3.3.2.1 三种制剂(F1、F4和RLD)
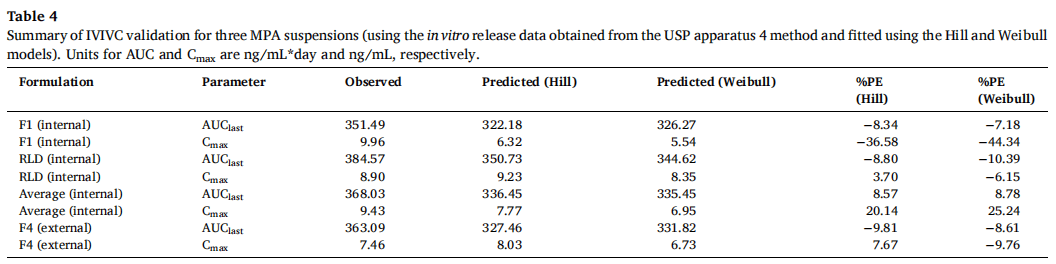
F1和RLD作为体内制剂,F4作为体外制剂,Hill和Weibull模型均较好地拟合了体外释放曲线,仅使用缩放方法就能建立和验证IVIVC。Hill和Weibull模型的IVIVC线性回归拟合系数(R2值)分别为0.9586和0.9599(图6A和6C)。通过建立的IVIVC对F4制剂的体内释放曲线进行预测和观察,除了曲线末端的几个时间点外均显示出良好的一致性 (图6B和6D)。采用Hill和Weibull模型,吸收比例因子(scaling factor for absorption)分别为0.91和0.93,时间比例因子(scaling factor for time)均为0.06。表4通过体外释放数据的Hill和Weibull模型对IVIVC验证进行了总结。对于不同的模型,IVIVC的预测性略有差异,比如使用Hill和Weibull模型,由于F1制剂Cmax的%PE绝对值较高,体内预测性是不准确的。但对于AUC和Cmax,体外预测性的%PE均在10%以内(Hill模型:AUC和Cmax的%PE分别为-9.81%和7.67%;Weibull模型:AUC和Cmax的%PE分别为- 8.61%和-9.76%)。根据FDA关于IVIVC缓释片口服剂型申请指南,当体内预测性不准确时,若体外预测性的AUC和Cmax的%PE绝对值均小于10%,可采用体外预测性进行IVIVC的评价。因此,使用F1、F4和RLD三种制剂成功建立了A级IVIVC,并使用USP装置四获得了体外释放数据。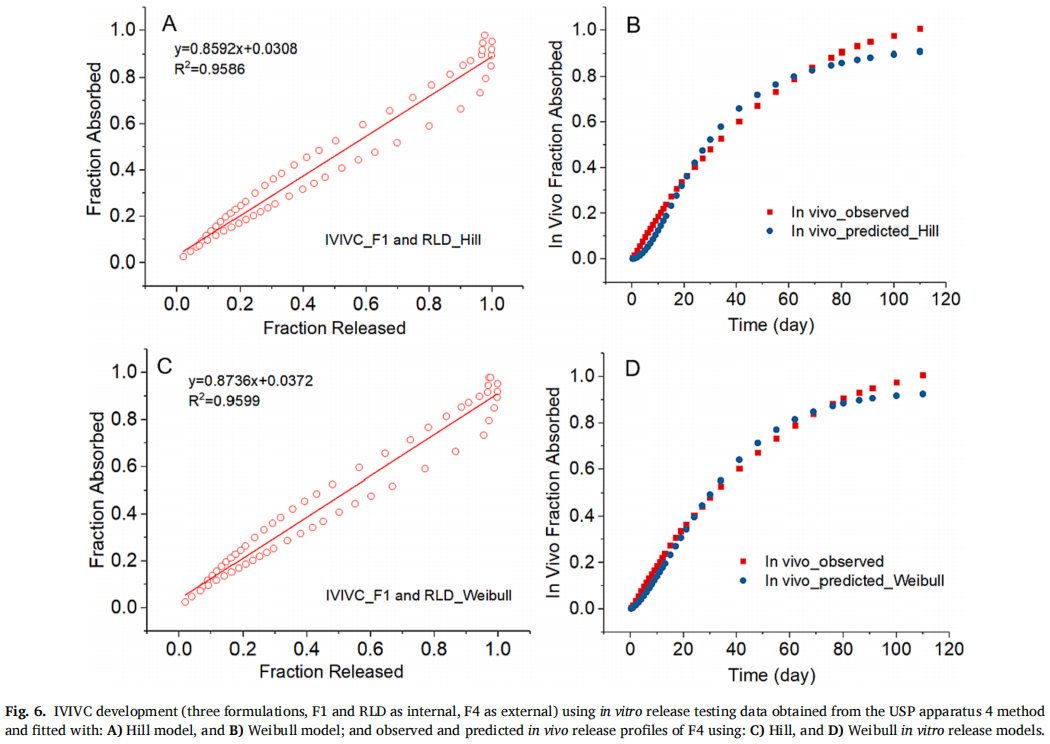
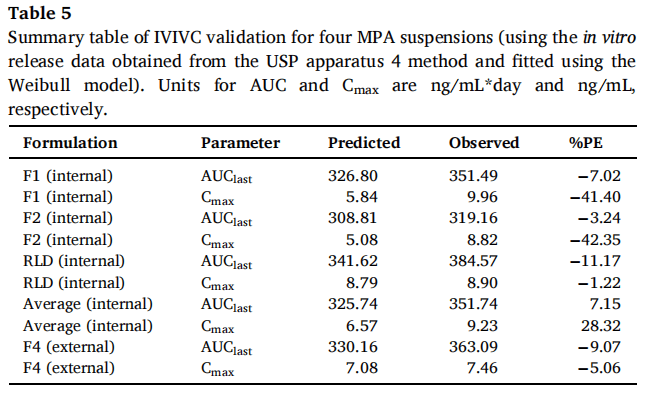
3.3.2.2 四种制剂(F1、F2、F4和RLD)
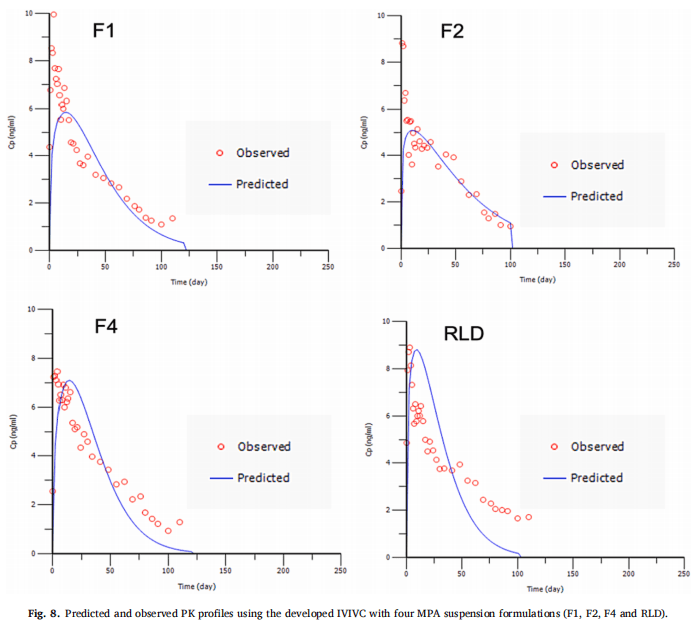
F1、F2和RLD作为体内制剂,F4作为体外制剂。使用Hill和Weibull模型均可获得拟合度高的体外释放曲线。但使用Hill模型拟合的体外数据获得IVIVC体外预测性的%PE值略超出标准(AUC的%PE值为-9.49%,Cmax的%PE值为12.26%)。因此,在IVIVC建立过程中选择Weibull模型进行体外释放数据进行建模,建立IVIVC并使用缩放方法进行验证。使用F1、F2和RLD,吸收和释放分数之间的线性回归拟合系数(R2值)为0.9664(图7A)。观察和由IVIVC预测的体内曲线在多数时间段有重叠(图7B)。吸收和时间比例因子分别为0.92和0.06。IVIVC验证总结如表5所示。与使用三种制剂建立的IVIVC类似,F1和F2 %PE绝对值较高,体内预测性不确定。但AUC(-9.07%)和Cmax(-5.06%)的%PE均在体外预测性的误差范围内(10%)。因此,使用四种MPA混悬剂和从USP装置四获得的体外释放数据成功建立了A级IVIVC,并且比较了四种制剂预测和观察到的PK曲线(图8)。由于Cmax的 %PE绝对值高,低估了F1和F2的Cmax预测值。相反,F4和RLD的Cmax预测值与观察到的Cmax显示出良好的一致性。由于目前关于长效注射混悬剂的IVIVC研究文献报道很少,这类制剂的Cmax中%PE较高的原因尚不清楚,预测误差较大可能是因为:(1)建模所用的数据为平均值,Cmax值可能发生偏移;(2)体外与体内释放条件有很大区别,而且要同时考虑到给药过程中对制剂产生的全部生理及生物学效应很难做到;(3)随着时间的推移,制剂可能在注射部位发生变化,例如沉淀、再结晶和聚集。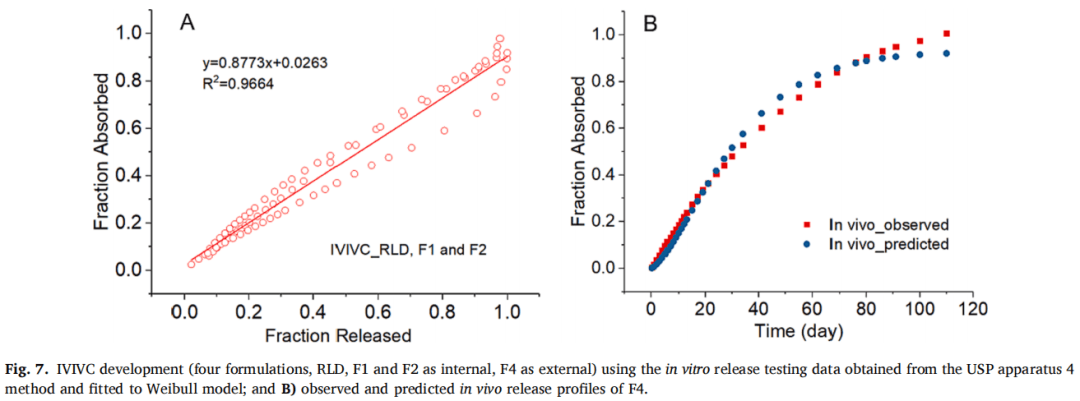
3.4 IVIVC研究的考虑因素
3.4.1 体外释放模型
本研究使用三种不同的模型(Hill、Weibull和双Weibull)进行体外释放曲线的数据分析。USP装置二获得的释放曲线在使用双Weibull模型时拟合最佳,而USP装置四获得的释放曲线在使用Hill和Weibull模型时拟合最佳。在通过三种制剂(F1、F4和RLD)研究IVIVC的过程中,使用USP装置四和Hill或Weibull模型成功建立了A级IVIVC。但通过四种制剂(F1、F2、F4和RLD)研究IVIVC时,只有使用Weibull模型拟合体外数据才能建立A级IVIVC,因此选择合适的模型对IVIVC研究至关重要。
3.4.2 体内释放数据和IVIVC研究
FDA指南并未规定建立IVIVC所需要的方法,只要所用模型在不同的IVIVC类别下具有可接受的%PE即可。通过IVIVC开发的长效注射混悬剂均能满足所有制剂的AUC标准(个体%PE<15%)。但是一些体内和体外制剂Cmax的%PE绝对值高,导致通过USP装置二无法建立A级IVIVC。相比之下,尽管USP装置四的体内预测性不准确,但仍成功建立了A级IVIVC。
这是关于长效注射混悬剂临床前(兔模型)IVIVC研究的第一份报告。通过USP装置四使用三种或四种制剂获得了体外释放数据,成功建立了A级IVIVC。由于制剂的Cmax的预测误差值较高,不能使用USP装置二获得的体外数据来建立IVIVC,因此在长效注射混悬剂的体外释放方法开发过程中,出于产品质量控制和IVIVC研究的目的,可以优先考虑USP装置四方法。药物粒径显著影响体外和体内释放过程,被认为是长效注射混悬剂开发中的一个关键质量属性。体外释放模型的选择(Hill、Weibull和双 Weibull等)可能会改变IVIVC建立的最终结果,因此在数据分析中必须仔细考虑。所建立的IVIVIC将为长效注射混悬剂的处方筛选和优化,以及体外释放方法研究提供科学依据。
略






