

时间:
 翻译:华溶应用中心
翻译:华溶应用中心
审核:工业药剂发烧友
一、介绍
自20世纪60年代以来,制药公司一直负责监控口服剂型在受控介质中溶出特性。早期溶出度检测集中于制剂制造的质量控制。溶出度测试提供了独特的监测综合生产参数的能力,影响溶出速度的有:片剂硬度,赋形剂控制,粒径等。该技术成为全球口服剂型常规检测所必需的。早期溶出度测试的历史记录在文献中。
早期溶出测试的另外的优点是活性药物成分(API)及其相关剂型的相对简单。这些可溶性药物易于生物利用,因为它们在胃肠道中往往是高溶解性的和高渗透性的。因此,对于高溶解性和高渗透性的药物,溶出度评估足以通过简单的标准溶液(如水和酸性介质)来确保临床表现。分歧主要侧重于活性成分行为可预测的固体口服剂型的崩解和分散。在这些条件下,溶出度测试在预测人类临床试验中的表现是有效的,并被表示为临床相关的方法。随着药物制剂技术的进一步发展,溶出测试在今天仍在使用的四种基本仪器上进行协调,通常由美国药典(USP)的1法,2法,3法和4法。其他USP仪器指定,例如7法,倾向于用于非口服剂型。
今天,溶出度测试扩大了其应用范围,并将其从简单的质量控制测试向前推广到用于预测体内/体外相关性(IVIVC)的可溶性和可渗透剂型以及许多其他临床相关性。在溶出测试的开发年代中发现的简单制剂,已经让位于更复杂的技术,以提高溶解性较低的分子的生物利用度。本书的目标是超越以前提及的现有溶出度文本,主要表示可溶性药物的溶出度测试,并且着重于目前分子在当前开发中的溶出度测试的问题。本文建立在以前的作品的坚实基础上,这些作品目前应用于溶出和药物释放技术,重点是难溶性药物。
二、改变药物重点
USP1-4的溶出平台是促进可溶性药物上市的关键技术。所使用的技术、仪器和简单的缓冲体系,理想地表征正在开发的剂型。然而,由于活性药物成分(API)变得越来越复杂(就胃肠道的溶解度和渗透性而言), 这些简单的药物释放机制并不与之相关。生物药剂学分类系统(BCS)由Amidon等开发,作为美国食品药品监督管理局(FDA)的指南,用于预测口服剂型肠道药物吸收。BCS系统已经成为基于药物溶解度和肠道渗透特性,对口服药物吸收进行分类和评估的金标准。
2.1 BCS分类系统
BCS 对药物的分类是基于这样的前提,即当药物溶解时,这个浓度可以穿过膜并与肠吸收相关。通过在人体胃肠道的代表性的各种条件和pH的体外化学测试建立胃肠道溶解性。药物的渗透性是基于最初的亲脂性测试,并在动物模型、组织研究、培养的上皮细胞如 Caco-2测试,并最终在人体(质量平衡,绝对生物利用度肠灌注测试等)中进一步研究。 随着药物溶解度和渗透性的建立,BCS系统将药物分为四类,如表1.1所示。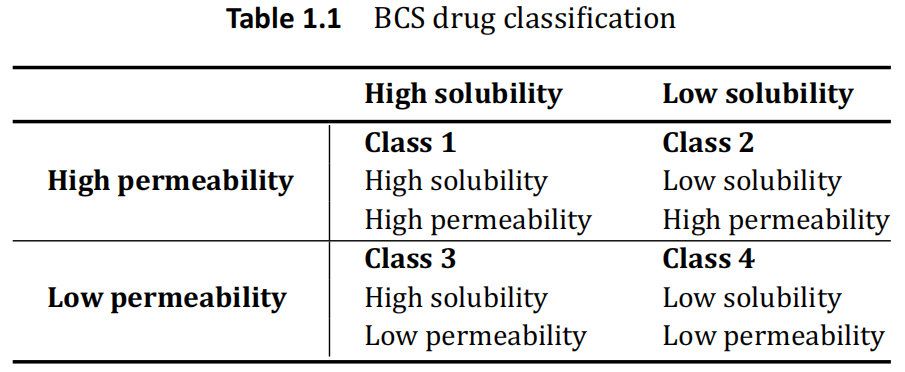
根据FDA指南,当在1至7.5的 pH范围内最高剂量规格可溶于<250mL水性介质时,目标药物被认为是“高溶解性的”。如果将人体吸收的程度确定为施用剂量的>90%,则该药物被认为是“高渗透性的”。此外,当使用USP 1或2,并在体积<500mL缓冲介质,在 15分钟内>85%标示量的药物溶出时,药物制剂被认为是“快速溶出”。
了解药物的 BSC类别允许制药科学家评估吸收药物的限速步骤。对于 BCS1类药物,API是高溶解度和高渗透性的,表明胃肠道中的吸收应溶出速率限制的。对于 BCS2类药物,由于药物溶解度较低但渗透性高,因此该类药物也应溶出速度受限。由于 3类溶解度较高,药物渗透性较低,因此该类API的吸收率受到限制。对于这类药物,溶出在临床上很少有相关性。FDA最近发布了一个指南,指出在特殊情况下,第3类药物可以按照生物豁免研究的1级规格进行处理。然而,本指南并没有提出通过传统溶出度测试,可以轻松实现 BCS 3类药物的 IVIVC。对于 BCS 1类和3类药物的IVIVC是不可能的,除非由于制剂(例如MR制剂)而导致药物溶出明显减慢,或化合物溶解度在 BCS1类的边界附近。FDA对速释固体口服剂型溶出度测试指南中承认,BCS4类药物依赖转运蛋白和其他生物手段在膜上运输。溶出可能能够表征这种运输,但是与 BCS 3类药物一样,溶出被用来表征这些药物的吸收。
2.2 难溶性药物
本书的一个主要重点是着重于 BCS 2类药物的溶出和药物释放。这些药物是高渗透性的,但溶解度有限。截至 2006年,BCS第2类药物占全球制药市场的三分之一。第5章将说明,制剂技术在提高这些分子的生物利用度方面已经有很大的进展。通常,这些制剂通常使用无定形或纳米颗粒材料技术。
三、溶出市场
溶出是在每个主要药物业务实验室中发现的口服剂型的重要技术,主要使用色谱和光谱进行最终定量分析。因此,或许是时候在本科仪器分析教科书中加入一章溶出度测试。2009年,约3400台仪器出售,市场预计年增长率为8%。今天,该市场价值超过1.5亿美元,直接与制药市场相关。制药业约占这些销售额的 75%。其余 25%分为合同研究机构,生物技术学院,学术界和农业部门。
如图1.1所示,质量控制测试主导了溶出度测试的需求。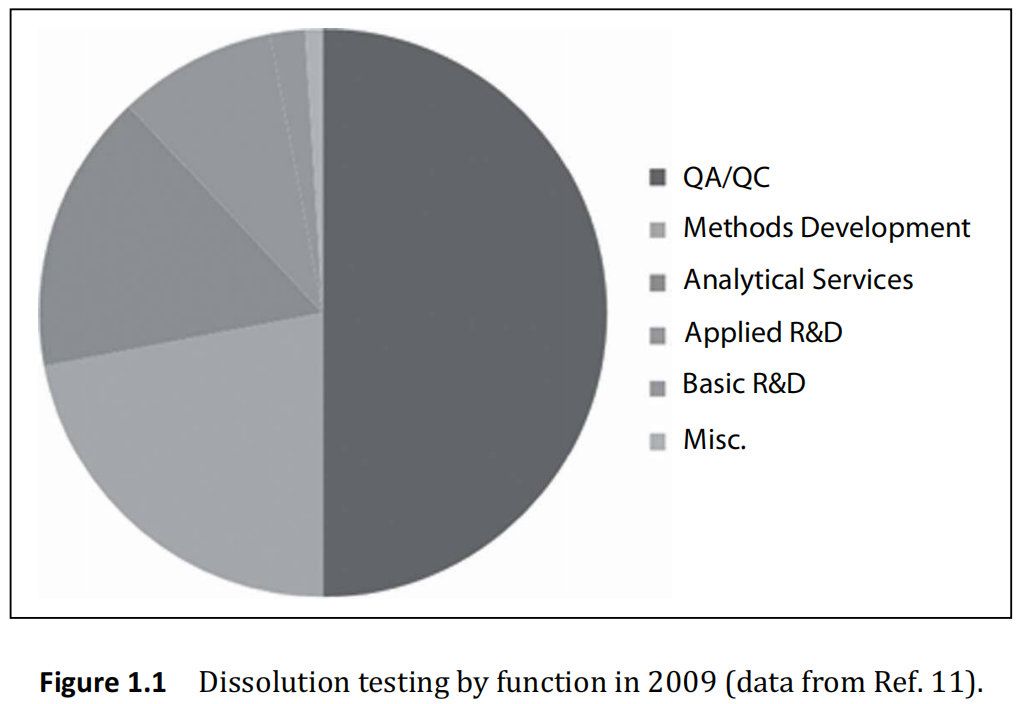
如图1.2所示, 2009年,最大的溶出供应商是 Varian/Agilent。供应商的市场多元化;然而,即使有这种多样性,今天的技术仍然围绕USP指定1法和2法的标准,市场上仪器3和4的部分不太重要。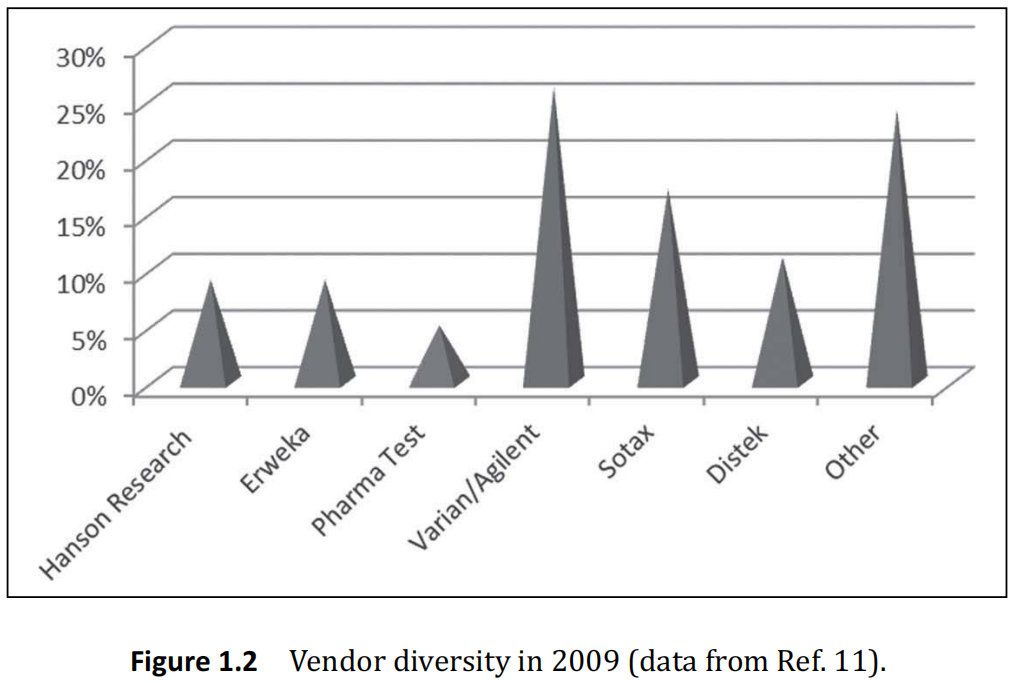
然而,正如第8章和第9章所述,这些仪器技术可能在未来几年的溶出和药物释放测试中发挥更重要的作用。
四、制药中的溶出及药物释放
本文的目的是为该领域领先的科学家讨论目前将溶出和药物释放应用于难溶性分子的应用。每一章都提供了所提供材料的主要从业人员的最新观点。尽管溶出度和药物释放的许多仪器并没有从传统的USP仪器上发生巨大的变化,但随着不断变化的分子被推向市场,这些方法和应用已经向前发展。本书建立在从最初和早期发展的技术上。有关溶出度和药物释放的发展和理论的细节可以在文献中找到。
4.1 药物 API 的溶解度测定
最终制剂中活性药物成分(API)的水溶性是影响药物的药代动力学特征的重要因素。有各种分析方法和预测 API溶解度的计算模型。在体内的溶解度,常常与通常用于药物质量控制过程的体外缓冲液体系中测定的不同。计算模型的质量也受实验溶解度数据的准确性影响。高通量发现过程推动了高通量筛选过程的开发,用于测量物理化学性质,包括脂溶性,pKa和溶解度,但是这些方法的准确性常常受到速度的要求和在测定中非生物相关介质的使用的影响。来自哈德斯菲尔德大学的 Kofi Asare-Addo和 Barbara R. Conway 将讨论关于 API的溶解度测定的细微差别。
4.2 在溶出度测试中使用表面活性剂
制剂的任何相关的溶出或药物释放测试之前,分析目标必须溶解在测试介质中。在溶出试验中,通常通过向溶出介质中加入表面活性剂来实现。表面活性剂的主要目标是在溶解后将目标分析物保持在溶液中。表面活性剂还通过降低介质中的表面张力来帮助材料润湿。然而,理想地,表面活性剂不应该通过加速药物制剂本身的溶解或侵蚀来改变释放的机制。Zydus的Amit Gupta 回顾了在溶出介质中使用表面活性剂,以将该技术扩展到难溶性药物。
4.3 难溶性药物的固有溶解速率评价
圣保罗大学的 Michele Georges Issa 和 Humberto Gomes Ferraz讨论了固有溶解度测定及其在难溶性药物评价中的应用。他们讨论了最常用的设备和固有溶解速率 (IDR)的计算,以及应用的一些相关实例和该方法的变量, 和实验设计的使用的简要说明。在药物化合物开发的所有阶段(从新化学实体的合成直到 API开发的质量保证) 中应用 IDR,科学家们可以更好地理解使用相对少量的材料来选择最佳生物利用度的主要候选参数的关键参数。
4.4 口服不良溶解药物的策略
在过去几十年中,组合化学和高通量筛选的发展,使得能够发现新的化学实体(NCE)以用于各种复杂和多样的生物学靶标。许多生物靶点本质上是高度亲脂性的或疏水性的,并且水溶性差的 NCE数量明显增加。虽然口服给药仍然是NCEs最常用的给药途径,但水溶性较差会导致明显的开发挑战,如吸收不完全,生物利用度高度变化,以及临床前物种和人类药物动力学差异大。现在可以使用许多常规和有利的制剂技术,来解决溶解性差以及由此导致的NCE的生物制药性能差的问题。然而,在发现过程中通常没有对最佳制剂技术进行系统的评估和主动选择, 而且由于生物制药不佳以及缺乏足够的临床前安全评估暴露,许多有希望的 NCEs被终止。本章的目标是描述一个系统的方法,用于生成一个交叉功能的数据包,包括物理化学,生物制药,ADME,PK/PD和输送技术评估,以口服递送水溶性差的 NCEs。Akash Jain, Dev Prasad 和 Sudhakar Garad 讨论了从临床前到商业开发中将可溶性分子配制成可行剂型的一般概念。
4.5 药物溶出度测试的分期方法
随着目标分子从发现到药物开发的各个阶段,溶出方法的应用也在不断发展。随着有关药物及其选定剂型的更多信息的了解,溶出方法性能的预期也随之增加。在临床开发的第2b/3阶段后期,溶出方法应该是其最终形式。对临床试验和生产历史的结果进行审查,以提出商业上市材料的相关规范。GregoryWebster, Paul Curry 和 Abb Vie 的Xi Shao 回顾了溶出方法的发展, 因为药物运输贯穿了药物开发的各个阶段。
4.6 两相体外开发与应用
包含水相和有机相的两相溶出测试被设计为比具有“吸收环境”的常规单相溶出测试更具生理学相关性。这是体外试验模拟药物溶出和分配到肠膜的动力学过程。本章报道的案例研究表明,与传统的单相溶出方法相比,这种两相溶出度试验为建立几种药物(包括速释和延长剂型)的体外和体内关系(IVIVR)提供了更好的机会。特别是,两相溶出试验可能非常有用地评估 BCSⅡ药物的亚稳态过饱和状态,包括过饱和的持续时间和程度,聚合沉淀抑制剂的作用以及药物分布的总体效应。另外,还讨论了重视药物分配动力学的两相溶出系统的理论模型。Abb Vie 的Gao Ping, Yi Shi 和Jon Miller 回顾了双相溶出在难溶性药物中的应用。
4.7 在难溶性药物制剂的溶出度测试中使用仪器3
USP3,往复筒最初设计用于测试延长释放产品并使药物制剂暴露于与胃肠道中发现的相似的药代动力学和机械性质。该仪器已被证明对难溶性化合物,咀嚼制剂,速释,延迟释放和许多改良释放产品有用。在写这篇文章的时候,往往需要对透析膜内的微球进行适应性评估,这样可以为生物相容性药物释放微粒和纳米粒子提供额外的好处。由于该仪器能够表征早期药物制剂候选物的释放特征,该装置有可能为通过质量源于设计(QbD)理念开发的制剂提供基于知识的评估,并进一步表征产品设计空间,用于批准后制造。
4.8 在难溶性溶解药物制剂的溶出度测试中使用仪器4
Sotax的 Geo ff rey N. Grove 与康涅狄格大学的 Rajan Jog和 Diane Burgess一起简要介绍了 USP 4技术的发展历程、药典考虑因素和监管考虑因素,以帮助为何时选择 USP4奠定基础。它还涵盖了各种不同系统配置和流通池选择和设计的概述,涵盖了药典和非药典设计选择。它进一步提出了诸如泵选择和流速等考虑因素的方法参数,并强调了可能与难溶药物相关的选择。
本章还对受检药物进行了全面的综述,包括文章标题,仪器和系统设计说明。总结了包括介质类型,过滤器类型,流量和各种其他参数在内的实验条件,重点在于选择 USP4的结果的相关性。作者希望对目前的文献进行回顾将是有用的参考和决策工具。
4.9 纳米颗粒药物制剂的溶出度
Shire LLC的 John Bullock 讨论了与纳米颗粒药物制剂的溶出相关的许多挑战中的一些。在过去几十年中已经开发了许多类型的基于纳米颗粒的药物制剂技术,以便为治疗和诊断剂提供一系列不同的增强的药物递送特性。在口服药物递送领域中,最常见的纳米颗粒结构可以分为纯药物纳米颗粒,其由基本上 100%的药物或各种不同的纳米尺寸结构组成,其中活性药物包封或分散在固体,半固体,或制剂基质内的液体状态。不管这两种类型的药物纳米颗粒中的哪一种以口服剂型使用,遇到的基本挑战和开发用于测量药物释放的合适技术的方法通常与纯药物纳米颗粒和基质型纳米颗粒制剂相似。这些纳米颗粒制剂的溶出方法的两个更明显的特征包括,在适当取样和处理含有小纳米尺寸药物颗粒的溶出样品中遇到的挑战,以及与用常规尺寸的药物颗粒制备的制剂相比具有更快的溶出速率的潜力。本综述首先简要讨论了控制药物颗粒的溶解度和溶出度的理论基础,以及对纳米尺寸药物颗粒更为重要的某些考虑,重点是纯药物纳米颗粒。在讨论之后,提供了引导从业者开发合适方法的实用指南,其中包括所使用的溶出介质的考虑因素,仪器考虑以及已被证明对许多不同类型的纳米颗粒制剂成功的取样和处理方法。
结合对仪器考虑的讨论,综述了最近出现的各种原位分析技术,并且证明了快速溶出的纳米颗粒制剂的溶出测试的优点,并且已经描述了用于评估纳米颗粒性能的一些替代的体外释放技术检查药物制剂。
4.10 基于脂质的药物制剂的溶出
来自Boehringer Ingelheim的Stephen Caffiero描述了基于脂质的药物制剂的一般性质及其溶出行为。用于体内吸收的药物可用性,可以通过溶解在胶体分散体中的药物的呈现来增强。越来越多的难溶性药物在亲脂性基质中配制,这个行业趋势已经导致了美国药典(USP)专家组的合作,这些专家组编写了通用章节(通用章节 USP <1094>液体填充胶囊溶出测试和相关质量属性),特别关注这些制剂的溶出度测试,还有相关议题,如胶囊破裂测试,乳剂液滴大小的确定和案例研究,将在此进行讨论。
4.11 稳定无定形药物制剂的溶出
Banner Life Sciences 的Justin Hughey 讨论了溶出测试在表征稳定化无定形药物制剂(SADF)中的关键作用。无定形系统在口服BCS 2类药物中继续是重要和不断增长的技术。使用高能形式的药物物质,例如无定形形式,与沉淀抑制性赋形剂相结合已被证明是增加溶解度和生物利用度的有价值的策略。本章解释了化学,物理形态,功能赋形剂,过饱和度和热力学的相互作用在溶出测试中如何融合,以深入了解与 SADF 相关的溶液介导的传质和相变现象。
在说明性案例研究中讨论了药物输送系统设计,溶出方法参数,介质选择以及漏槽和非漏槽条件的细微差别等基本考虑因素。这些研究清楚地表明了溶出和沉淀测试在理解和推进这种重要的药物递送方法方面的力量。
4.12 混悬制剂的溶出
混悬剂由分散于液体介质中的不溶性固体颗粒组成。开发混悬剂的最常见原因是活性药物成分(API)在所需剂量下的水溶性是有限的。使用混悬的另一个常见原因是与溶液相比,它们通常提供改善的化学稳定性。混悬剂还提供了味道掩蔽和某些患者(例如,小儿科)的更方便的剂型的优点。在本章中,辉瑞公司的 Michele Xuemei Guo, Kenneth Norris和 Ling Zhang着重于通过口服给药途径给药的剂型中的混悬制剂的溶出和药物释放,包括口服混悬剂,用于重建的混悬液和胶囊中的混悬液。
4.13 生物相关溶出
辉瑞公司的Mark McAllister和Irena Tomaszewska 在生物相关的溶出条件下讨论测试难溶性药物。一般来说,用于改善溶出度测试的生理相关性的方法可以大致分为两组。第一组包括机械方法,其中控制溶出过程的单个方面以研究诸如介质或流体动力学的生理变量的影响。第二组包括设计用于模拟胃肠道多个方面的方法和设备,并提供包含流体,消化,转运和吸收的腔内条件的整体模拟。应当认识到,对于剂型或API溶出的特定方面,机械(还原剂)方法可以允许详细评估个体现象,例如过饱和度,沉淀和再溶解。相比之下,整体模拟方法考虑了多个过程(如消化,转运和吸收)对剂型表现的总和或净效应,例如在评估复杂的食物效应时。本章介绍了机械和整体方法的生物相关溶出测试的开发,并评估了在溶出测试中对介质(组成和体积),流体动力学和吸收组分的整合修饰的生物相关性。
4.14 低溶解度速释产品的临床相关溶出度
AstraZeneca的Paul Dickinson, Talia Flanagan, David Holt 和Paul Stott讨论了低溶解度速释产品的临床相关溶出情况。他们提出了一个结构化的开发方法,努力确定和评估临床表现的风险,并在适当的情况下,测试体内这些风险的影响。每一种化合物都应该根据其自身的优点加以考虑,但是采用提议的5步法将确保考虑所有重要的因素, 并最终导致建立一个强有力的控制策略。这种方法的好处包括增强产品供应的安全性,优化制造工艺的能力,并展示任何提议的变更的(缺乏)影响,以及改善对患者提供的产品临床质量的保证。预期溶出度测试及其相关规范可以用于许多目的,因为区分力的要求和完全释放的演示可能会发生冲突。如果我们要实现更具临床相关性的溶出指标的全面好处,就需要进一步开展科学的理解和监管协调工作。作者们相信,在 QbD的支持下提出的增加知识和理解的开发是基于对体内表现的洞察和控制。
4.15 BCSⅡ/Ⅳ类产品溶出度测定方法验证和 QbD
葛兰素史克公司的 Alger Salt 讨论了 QbD方法开发和验证溶出度测试方法。QbD原则为产品、技术和流程的开发和交付提供了结构化的方法。历史上,QbD一直针对制造工艺和产品属性。QbD原则现在正在应用于分析方法。方法转移是成功的产品生命周期的一部分,因为R&D中开发的工作最终被转移到制造场所的QC 实验室。
在采纳 QbD原则之前,分析人员学习(并多次重新学习)分析方法的问题经常在方法转移过程中发现。现在不是发现这种问题的好时机,因为通常很难对方法进行修改,修复这些问题可能令人沮丧,而且费用昂贵。
1.设计意图
2.设计选择
3.控制定义
4.控制验证
4.16 BCSⅡ类/Ⅳ类产品的药物释放测试中的监管注意事项
药物与生物技术开发的 Robert Bell 和武田药业的 Laila Kott 总结了目前的指导原则,以及它们与低溶解度化合物的关系。我们回顾仪器的历史和进展,以及指导文件的演变。介绍了用于描述所有类型化合物的三个分类系统的总结。分类系统包括 1995年出版的 1995年药物产品生物药剂学分类系统(BCS), 2005年出版的生物药剂处置分类系统(BDDCS)和2010年首次提出的可开发性分类系统(DCS)。
介绍了关于使用生物相关介质进行制剂开发的适用性及其用于质量控制(QC) 测试的可能性的讨论。讨论和剖析了关于这一主题的指导原则。其中包括与目前的指导意见和行业趋势有关的溶出指标的设定。最后,总结了当前的关键指导文件和新兴的监管议题。
4.17 胶囊制剂的溶出度
配制在液体填充胶囊中用于吸收的化合物的可用性,取决于胶囊壳的初始溶解和破裂,并随后其填充内容物在 GIT 液体中的释放和溶出。这两个过程需要在释放时和胶囊产品的保质期内进行监测。由于壳体和填充材料的复杂性质,液体填充胶囊在开发和应用溶出方法期间提出了独特的挑战。外壳材料容易发生其机械性能的变化或明胶的交联,这导致其溶解度的变化。另一方面,填充材料可以在混悬液填充物中,显示出悬浮物质的粒度分布或多晶型性质,或溶解填充物中的可溶性化合物的结晶的变化。在后一种情况下,溶解的化合物的结晶,可以在胶囊剂型,或在填充材料遇到体外和体内含水流体时发生。
溶出度测试是体外表征液体填充胶囊产品的非常有价值的工具,常规用于
(a)评估批次间质量,
(b)监测产品在其保质期内的质量变化,
(c)在扩大规模和批准后变更(SUPAC)后评估产品相同性,
(d)符合生物豁免要求, 降低产品规格;
(e)符合生物豁免要求,用于在本地采取行动的产品 GIT。
此外,设计用于产生体外/体内相关性(IVIVC)或体外/体内关系(IVIVR)的溶出方法可用于预测产物之间潜在的生物等效性或生物不等效性。Dart Neurosience LLC的 Rampurn a Gullapalli写的这一章的目的,是对影响液体填充胶囊产品的体外和体内溶出的因素进行深入讨论,开发其常规QC 测试的溶出方法,以及对这些方法产生潜在的 IVIVC和IVIVR。
4.18新兴和非药典释放技术
常规和非常规剂型的溶出度检测是在二十世纪六十年代引入的,现在作为一项必不可少的药典试验,由所有药典用于评估药物释放。除了标准化测试外,还有几种非药典方法被合格和验证用于药物释放测试。
本章分为三个部分。第一部分简要讨论现有的药典方法以及任何非药典方法。第二部分描述了不同类型剂型的非临床仪器。最后一节将介绍各种检测技术,如使用拉曼光谱, FTIR-ATR和光纤的 UV成像。康涅狄格大学的 Namita Tipnis 和Diane Burgess提供他们对药物实验室溶出和药物释放测试的新兴技术评估的回顾。
五、参考文献
略
如需原文,请联系小编(15012941165)






