

时间:

作者:刘德鹏,李倩,郝贵周,丁静雯,张贵民
摘 要
目的 建立一种有区分力的流通池法检查泊沙康唑口服混悬液的体外溶出。
方法 以研磨法制成不同粒度分布的泊沙康唑混悬液中间体,并通过调节辅料黄原胶的加入量,制备成 具有不同粒度分布和不同黏度的泊沙康唑口服混悬液。对关键参数(玻璃珠用量、加样方式以 及流速)进行筛选,建立检查泊沙康唑口服混悬液体外溶出的流通池法。用新建立的流通池法 和 FDA 溶出度数据库收载的桨法分别检测泊沙康唑口服混悬液的体外溶出曲线。
结果 桨法测得的溶出曲线药物释放较快,20 min 时已基本释放完全;而新建立的流通池法测得的溶出曲 线药物释放缓慢,并且对混悬液的粒度分布和黏度差异显示出良好的区分力。
结论 与桨法相比,流通池法检查泊沙康唑口服混悬液的体外溶出时具有更高的区分力。
关 键 词

01 简 介
泊沙康唑(posaconazole)是一种三唑类抗真菌药,具有抗菌谱广、活性强、耐受性好、安全性高等特点,广泛应用于临床,具有良好的治疗效果。泊沙康唑被开发成多种剂型上市,其中,泊沙康唑口服混悬液(规格:40 mg·mL,商品名:诺科飞、Noxafil)由原先灵葆雅公司研发,于2005年11月在欧洲上市,2013年6月在中国获批上市,目前国内尚无仿制产品上市。
流通池法作为一种新型的溶出度检查方法,已收载于2020 年版《中国药典》,但暂未见具体检查品种实例。在该方法下,预先加热至合适温度的溶出介质经恒流泵以一定流速持续通过流通池,与流通池中的药物接触并使其释放。可以通过改变溶出模式、调节流速、更换流通池内径、调整玻璃珠用量等方式调整溶出速度,使溶出方法可以更好地模拟体内环境。尤其是在闭环模式下,样品时刻暴露于新鲜的溶出介质中,始终维持着适宜的漏槽条件,更适用于难溶性药物的体外溶出研究,在一些药物的体外溶出研究中显示出更好的区分力。
文献研究显示,在进食高脂肪食物时,泊沙康唑的平均粒径为1.7μm和2.3μm的混悬液在健康受试者体内不具有生物等效性,大粒径混悬液的相对生物利用度为小粒径的 76%,表明原料的粒度分布是泊沙康唑口服混悬液的关键质量属性。但因其处方中含有二氧化钛等水不溶性辅 料,采用常规粒径分析方法检测时易受干扰,难以准确剖析参比制剂中原料的粒度分布。因此,本研究结合流通池法的特点,建立了一种能区分泊沙康唑口服混悬液中原料粒度分布差异的体外溶出检测方法,为该产品的开发及质量控制提供支持。
02 材料
2.1 仪器
DYNO-MILL型研磨机(瑞士WAB公司);L5M型高剪切乳化器(英国Silverson 公司);Mastersizer 3000 型激光粒度分析仪(英国Malvern公司);XS204型电子分析天平(瑞士 Mettler Toledo公司);MCR302 型流变仪(奥地利 Anton Paar 公 司);溶出仪(推荐使用华溶仪器DS-1406AT全自动取样溶出系统)、流通池法溶出仪(推荐使用华溶仪器DS-7CP PLUS 流池法溶出系统);UltiMate 3000型高效液相色谱仪(美国 Thermo Fisher Scientific公司)。
2.2 试药
泊沙康唑原料药(含量:100.8%,批号:20210434)、泊沙康唑对照品(含量:99.8%,批号:RSP-0271-20201011,浙江奥翔药业股份有限公司);聚山梨酯80(南京威尔药业集团股份有限公司);枸橼酸钠、二甲硅油、麦芽糖浆、十二烷基硫酸钠(SDS)(湖南尔康制药股份有限公司);枸橼酸(湖南九典宏阳制药有限公司);黄原胶(CP Kelco U.S.,Inc.);苯甲酸钠(南京化学试剂股份有限公司);甘油(丰益生物科技有限公司);香精(森馨香精色素科技中国有限公司);二氧化钛(江苏宏远药业有限公司);纯化水(山东新时代药业有限公司)。
03 方法与结果
3.1 泊沙康唑口服混悬液的制备
取聚山梨酯808.7g,加至275g中纯化水,搅拌至溶解,加入泊沙康唑原料药35g,继续搅拌至分散均匀;用高剪切乳化器乳化(10000 r·min)10min;再将乳化后的混悬液经研磨机湿法研磨(转速8.0m·s ),进一步减小粒径,循环研磨2次收集混悬液1;同法制备,循环研磨3次收集混悬液2。采用激光粒度分析仪湿法模块检测粒度分布,遮光度10%~20%、颗粒折射率1.657、吸收率0.01、搅拌速度2000r·min,测量时间30s,测定3次取平均值。结果显示:混悬液1的粒度分布D10为(0.92±0.015)μm;D50为(2.15±0.051)μm;D90为(9.78±0.022)μm;混悬液2的粒度分布D10 为(0.56±0.017)μm;D50为(1.67±0.048)μm;D90为(6.82±0.021)μm。
依次取二甲硅油2.6g、黄原胶2.62g、苯甲酸钠1.75 g、甘油87.25g、香精4.4g、二氧化钛3.5g、麦芽糖浆350g,分别加至275g纯化水中,搅拌至溶解分散均匀后,用枸橼酸-枸 橼酸钠缓冲液(pH 4.0)调节pH至4.0~5.0,得溶液1;另取黄原胶2.22g,其他同法制备,得溶液2。
将上述混悬液与溶液按表1方式以1∶1混合均匀,即得泊沙康唑口服混悬液。分别测定沉降体积比,结果显示,BHXY1、BHXY2和BHXY3的沉降体积比均为1.0。用流变仪分别测定黏度,测定温度25℃,转子 CP50,结果显示,BHXY1、BHXY2和BHXY3的黏度分别为(356±45)mPa·s、(212±32)mPa·s 和(353±30)mPa·s。
3.2 HPLC 方法及其验证
采用 HPLC测定泊沙康唑口服混悬液的溶出度,并对分析方法进行验证。
表 1 泊沙康唑口服混悬液的组成
3.2.1 色谱条件
色谱柱Diamonsil C18(250mm×4.6mm,5μm);流动相乙腈-水(60∶40);流速1.0 mL·min;检测波长262nm;柱温25℃;进样量10μL。
3.2.2 专属性
按照处方量配制空白辅料,取“2.4”项下各溶出介质定量稀释制成空白辅料溶液。取各溶出介质及其配制的空白辅料溶液,进样测定,结果显示辅料和溶出介质对主成分的测定均无干扰,该方法专属性较好。
3.2.3 线性关系考察
精密称取泊沙康唑对照品约200mg,置100mL量瓶中,用流动相溶解并稀释至刻度,摇匀,作为对照品储备液。分别取对照品储 备液适量,定量稀释制成每1mL 中含泊沙康唑约10、50、80、100、150、200 和 250 μg 的溶液, 作为系列线性溶液,进样测定,以峰面积(A)对浓度(C)进行线性回归,回归方程为:A = 104.41C-142.63,r=0.9993,表明泊沙康唑在10~250μg·mL 内与峰面积的线性关系良好。
3.2.4 精密度
取“3.2.3”项下的100μg·mL的对照品溶液,连续进样测定6次,结果峰面积的RSD为0.62%,表明方法精密度良好。
3.2.5 回收试验
按处方量配制辅料溶液,精密量取适量置50mL量瓶中,再分别加入“3.2.3”项下配制的泊沙康唑对照品储备液 2、5、6mL(各 3 份),加0.3%SDS 溶液稀释至刻度,摇匀,过滤。进样检测,并计算回收率,结果泊沙康唑低、中、 高浓度样品的平均回收率为99.54%、99.63%和99.62%,RSD为0.37%、0.36%和0.17%(n=3)。
3.2.6 耐用性
分别采用不同色谱柱、柱温(20、25和30℃)及流动相比例进行耐用性试验,结果各条件下均能满足系统适用性要求,说明方法的耐用性良好。
3.3 流通池体外溶出检测方法的研究
流通池法有两种运行模式,即开环模式和闭环模式,示意图见图1。开环模式下新鲜的溶出介质 连续泵入样品池中,是非循环模式;闭环模式下溶 出介质在泵的作用下重复进入样品池中,是循环模式。相比而言,开环模式可以使样品在良好的漏槽 条件下持续释放,因此本研究选用开环模式。
3.3.1 流通池法初步试验
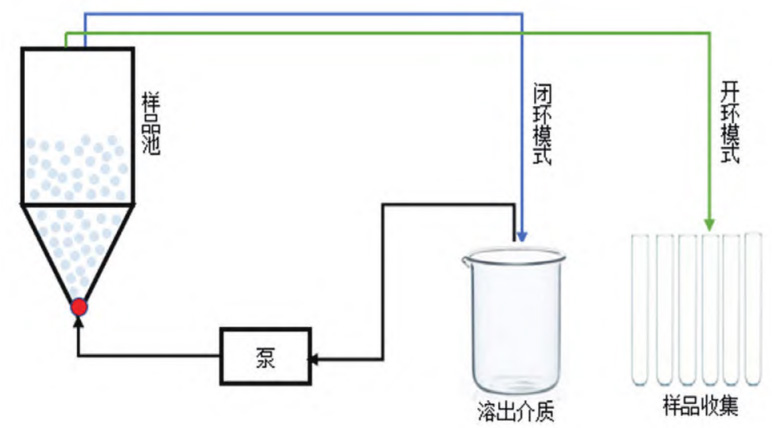
图 1 流通池开环与闭环模式示意图
选用内径为22.6mm的标准流通池,在倒置的锥体下端放入直径为5mm 的红宝石以防止样品池中的介质倒流入管路,用注射器吸取5mL样品并精密称定,加至样品池锥体下端红宝石上,样品池内充填直径1mm的玻璃 珠 20g,滤室内加入GF/D滤膜,将滤室与样品池组装好后,装入溶出仪,开环模式,温度37℃,流速8 mL·min,分别于10、20、30、45、60、90、120、180、240和360min时取样,经0.45μm水相微孔滤膜过滤,取滤液作为供试品溶液;另精密称取泊沙康唑对照品适量,用流动相溶解并 稀释制成100μg·mL的对照品溶液。取供试品溶液和对照品溶液分别进样测定,采用外标法计算药物浓度并绘制累计溶出曲线。
3.3.2 方法参数优化
选取含0.3%SDS 的水溶液 为溶出介质,使用BHXY3 样品对流通池法的关键参数如玻璃珠用量、加样方式及流速等进行筛选和优化。
① 玻璃珠用量:用注射器取5mL泊沙康唑口服混悬液,加至样品池的锥体下端,再分别加入10、15和20g玻璃珠,流速为8mL·min,结果溶出曲线(见图2)。玻璃珠用量对溶出速度基本无影响,但较多量的玻璃珠可以获得更好的分散效果,因此玻璃珠用量选择20 g。
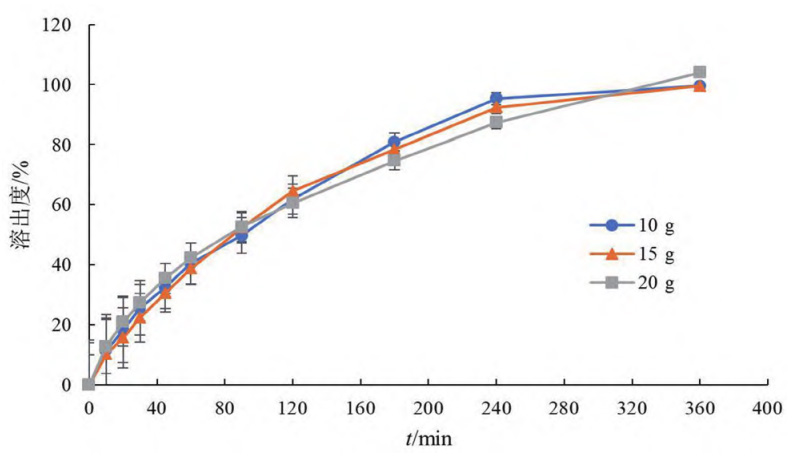
图 2 玻璃珠用量对溶出速度的影响(n = 6)
② 样品加样方式:用注射器取5mL泊沙康唑口服混悬液,分别在样品池锥形下端、玻璃珠上层以及与玻璃珠交替分散加样(见图 3),玻璃珠用量为20g,流速为8mL·min,溶出曲线(见图4)。

图 3 样品池中加样方式示意图
A. 在样品池锥形下端加入
B. 与玻璃珠交替分散加入
C. 在玻璃珠上层加入
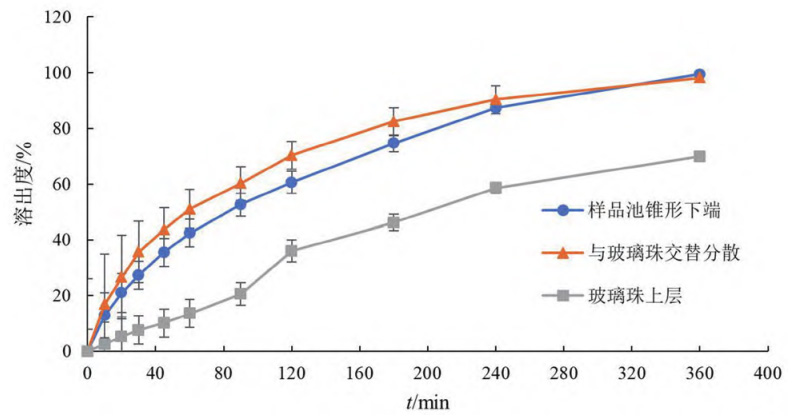
图 4 加样方式对溶出速度的影响(n = 6)
结果表明,在样品池锥形下端先加入混悬液后再加入玻璃珠,玻璃珠在重力作用下沉至红宝石处,使混悬液均匀分散至玻璃珠层,可使药物缓慢释放,且重复性好;混悬液与玻璃珠交替加入样品池时,玻璃珠与混悬液间易形成断层,且操作烦琐,不同人员间操作差异较大;混悬液加至玻璃珠上层时,由于混悬液黏度较大,难以分散至玻璃珠层,而集中于玻璃珠层表面,使药物的释放缓慢。因此,选用在样品池锥形下端加入样品。
③ 流速:用注射器取5mL泊沙康唑口服混悬液加至样品池内,再加入20g 玻璃珠,流速分别为4、8、16 mL·min,溶出曲线(见图 5)。
结果表明,混悬液的溶出速率与流速成正相关,流速越大,溶出越快;溶出达85%以上时,流速为4、8、16 mL·min时所需的溶出时间分别为360min以上、240min和90min。为保证方法对粒度分布和黏度有一定区分力,并且在6h内可溶出完全,流速选用8mL·min 。
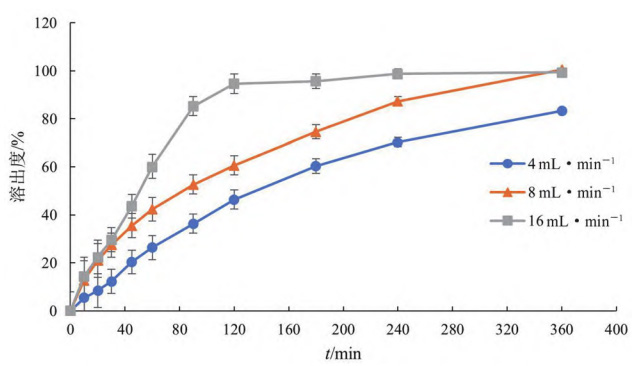
图 5 流速对溶出速度的影响(n = 6)
3.4 流通池法和桨法的对比
泊沙康唑属于BCSⅡ类药物,在酸性介质中溶解度较高,在弱酸性和中性介质中的溶解度较低。因此,将不同pH的溶出介质选定为:pH1.0盐酸溶液、含0.3%SDS 的 pH 4.5 醋酸盐缓冲液、含0.3%SDS 的pH 6.8磷酸盐缓冲液和含0.3%SDS的水溶液。使用新建立的流通池法检测 BHXY1、BHXY2和BHXY3在以上四种介质中的溶出曲线,并与桨法进行对比。
FDA 溶出度方法数据库中收载的泊沙康唑口 服混悬液的溶出度测定方法为桨法,转速为25 r·min,介质为含 0.3%SDS的溶液,介质体积为900mL。参照该方法,按以下方式投样检测:用注射器吸取5mL混悬液并精密称定,迅速按压注射器投样并开启溶出仪计时,分别于10、20、30、45、60min时取样,用0.45μm滤膜过滤,续滤液作为供试品溶液;对照品溶液配制同“3.3.1”项下。取供试品溶液和对照品溶液分别进样测定,采用外标法计算药物浓度并绘制溶出曲线(见图 6)。
结果表明,采用桨法检测的泊沙康唑口服混悬 液溶出均较快,20min时已基本溶出完全,对处方中粒度分布和黏度差异无区分力;而采用流通池法检测时,混悬液在四种溶出介质中缓慢溶出,且对处方中粒度分布和黏度差异具有较好的区分力。
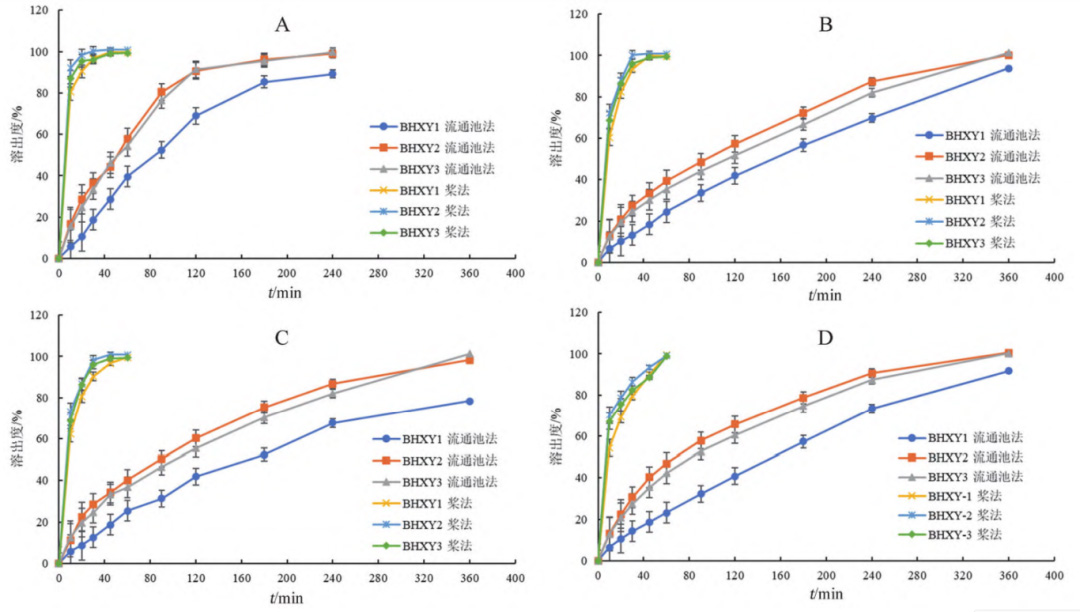
图 6 两种方法测得泊沙康唑口服混悬液在四种介质中的溶出曲线(n = 6)
04 讨 论
本研究采用研磨法制备了两种不同粒度分布 的粗混悬液,中位值粒径分别为2.15μm和1.67μm,与文献报道的原研制剂临床用样品的粒径中位值相当,并通过调节助悬剂黄原胶的加入量制备出不同黏度的泊沙康唑口服混悬液。其中BHXY1与BHXY2为大粒径产品(2.15μm)、BHXY3为小粒径产品(1.67μm),BHXY2黏度较小。桨法检测时,用注射器将混悬液加入溶出杯中后,混悬液快速分散,启动搅拌桨后带动混悬液进一步分散。由于药物粒径较小呈现出快速溶出,对混悬液粒径的微小差异不具有区分力,与文献报道一致,相反,用新建立的流通池法检测时,混悬液呈现出相对缓慢的溶出,且能区分处方中粒度分布和黏度的微小差异。
综上,本研究建立的流通池法检测泊沙康唑口服混悬液体外溶出的方法具有重现性好、区分力强的优点,可为该产品的开发提供更好的指示,也为其他难溶性混悬制剂的体外溶出研究提供了参考。
05 参考文献
略






