

时间:

作者:范诗雨,刘芳伶,贺劲杰,周群,程泽能
摘 要
目的 通过流通池法模拟富马酸喹硫平缓释制剂的体内动力学过程,建立具有体内外相关性的溶出方法,并进行体内外相关性模型预测能力的评估。
方法 采用HPLC-MS/MS 法测得T1、T2 和R制剂的血药浓度数据,通过WinNonlin软件的非房室模型和反卷积模块解析参比制剂的体内特征,依据参比制剂的体内特征指导溶出方法的设计,用3 种释放速率不同的制剂的体内和体外数据建立体内外相关性模型,并进行模型预测能力验证。
结果 建立了具有A级相关性(R²=0.967)和C级相关性(R²=0.996)的溶出方法,内部制剂预测和外部制剂预测的预测误差PE%<10%,符合标准。
结论 本方法具有良好的体内外相关性,其体内外相关性模型经验证预测误差符合标准,对于吸收速率不同的制剂均具有良好的区分力,可以为富马酸喹硫平缓释片的制剂评价提供参考。
关 键 词

01 简 介
富马酸喹硫平是英国AstraZeneca制药公司新开发的第二代非典型抗精神病药(second genera-tion antipsychotics,SGA)。其缓释制剂可以延长药物治疗作用持续时间,降低毒副作用,减少用药次数,改善用药的依从性。该药物于2013年在中国上市,由于其仿制难度较大,目前国内仅有3家仿制制剂为在售状态。体内外相关性(in vitro-in vivo correlation,IVIVC)模型是一种用于描述口服剂型体外特性(药物溶解或释放的程度)与体内性能(血浆药物的溶度和吸收量)之间相关性的数学模型。依据IVIVC模型对药物体内时间-血浆药物浓度曲线的拟合程度,FDA指南将IVIVC划分为A、B、C和多重C级4个级别。
本研究以富马酸喹硫平缓释片为研究对象,依据体内药动学特征来设计体外溶出方法,并初步建立和评估了该药物的体内外相关性模型。基于新型流通池法建立富马酸喹硫平缓释片具有体内外相关性的溶出方法,依据药物体外溶出行为来模拟预测药物的体内性能,这不仅有助于提高该制剂的生物等效性试验的成功率、节约仿制药研发成本、节省注册时间,还可以被用于建立体外溶出度标准,进行药品批间质量控制等。
02 仪器与试药
全自动流通池型溶出仪(推荐使用华溶仪器DS-7CP PLUS 流池法溶出系统);高效液相色谱仪(Agilent1260,配有 DAD 检测器);UX6200H 型百分之一电子天平 [岛津企业管理(中国)有限公司 ];十万分之一电子天平[赛多利斯科学仪器(北京)有限公司 ];HH-6 型电热恒温水浴锅(天津市泰斯特仪器有限公司);Vortex-5型可调式涡旋混合器(山海嘉鹏科技有限公司);TG20-WS 台式低速离心机(湖南迈克尔实验仪器有限公司);FE28 型 pH 计[梅特勒-托利多国际贸易(上海)有限公司],Eco-S15UVFV型实验室纯水系统(上海和泰仪器有限公司)。
受试制剂T1、T2(某公司自研产品,规格:200mg);受试制剂T3(某市售制剂,规格:200 mg);受试制剂T4(某市售制剂,规格:200mg);富马酸喹硫平缓释片(R,阿斯利康制药有限公司,批号:MM2654、MK2374);富马酸喹硫平原料药(浙江苏泊尔制药有限公司,批号:011-180305U),磷酸二氢钾(上海沪试,批号:20220110),氢氧化钠(上海沪试,批号:20210107),磷酸(天津市科密欧化学试剂有限公司,批号:20210710);三乙胺(广州光华科技股份有限公司,批号:20200809);盐酸(成都市科隆化学品有限公司,批号:2020112502);富马酸喹硫平对照品(纯度:96.2%,批号:2413-025A2)、富马酸喹硫平内标(纯度:95.7%,批号:3298-096A6)(TLC pharmaceutical Standards);甲醇、乙腈、乙腈均为色谱纯。
03 方法与结果
3.1 药物体内试验
3.1.1 T1与R的人体生物等效性试验
采用随机、开放、两制剂、两周期(T-R、R-T)、自身交叉试验设计,周期间的洗脱期为7d。试验方案经海口市人民医院生物医学伦理委员会 [ 伦理批准号:2021-(伦审)-172] 批准,受试者均签署知情同意书。48 例受试者在试验当日早上空腹口服 T1 或 R 一片,于给药前(0 h)及给药后 0.5、 1、2、3、4、5、6、7、8、9、10、11、12、13、 14、15、16、18、20、24、28、32、36、48 h 分别采集静脉血 4 mL,血样置于肝素钠抗凝剂真空采血管中,1h 内离心分离血浆,血样放于超低温冰箱(低于- 60 ℃)中储存待分析,采用高效液相- 质谱联用(HPLC-MS/MS)测定人体血浆喹硫平浓度,采用 WinNonlin软件非房室模块计算富马酸喹硫平的药动学参数。
3.1.2 T2与R的人体生物等效性试验
采用随机、开放、两制剂、两周期(T-R、R-T)、自身交叉试验设计,周期间的洗脱期为7d。试验方案经南华大学附属第二医院药物临床试验伦理委员会[ 伦理批准号:【202106-03】药伦审字(191)号]批准,受试者均签署知情同意书。48例受试者在试验当日早上空腹口服T2或R一片,于给药前(0h)及给药后 0.5、1、2、3、4、5、6、7、8、 9、10、11、12、13、14、15、16、18、20、24、 28、32、36、48h分别采集静脉血4mL,血样置于肝素钠抗凝剂真空采血管中,1h内离心分离血浆,血样放于超低温冰箱(低于-60 ℃)中储存待分析,采用HPLC-MS/MS测定人体血浆喹硫平浓度。
3.1.3 生物样本检测
① 色谱条件。
色谱柱:Waters XBridge C18(4.6mm×50mm,3.5μm),流速:0.6 mL·min;进样量:10µL;柱温:35℃;流动相:0.1%甲酸水溶液为A泵,甲醇为B泵;梯度洗脱:0.00~1.00 min,80%A;1.00~1.5min,80%→10%A;1.5~4.5 min,10%A;4.5~4.6min,10%→80%A。
② 质谱条件。
离子源:电喷雾离子源ESI;检测方式:正离子MRM 模式;离子源喷雾电压:4000V;离子源温度:300 ℃;碰撞能:喹硫平内标和喹硫平的碰撞能均为25V;监测离子对:喹硫平内标的检测离子为m/z 392.2~258.1,喹硫平的检测离子为m/z 384.2~253.1。
HPLC-MS/MS法测得各制剂单位时间的平均血药浓度检测结果见图1a、图1b,将各制剂的血药浓度 -时间曲线数据代入WinNonlin 软件中的反卷积模块可求得各制剂的各时间点的吸收百分数图(见图1c),各制剂的吸收速度和吸收程度差异明显,T1>R>T2。采用非房室模块计算富马酸喹硫平的药动学参数,比较T1、T2和R制剂的Cmax、AUC0~t和AUC0~∞的几何均值比Ratio(T2/R)和Ratio(T1/R),结果也说明在体内试验中T1的吸收程度比R高、吸收速度比R快,T2的吸收程度比R低、吸收速度比R慢(见表1、2)。
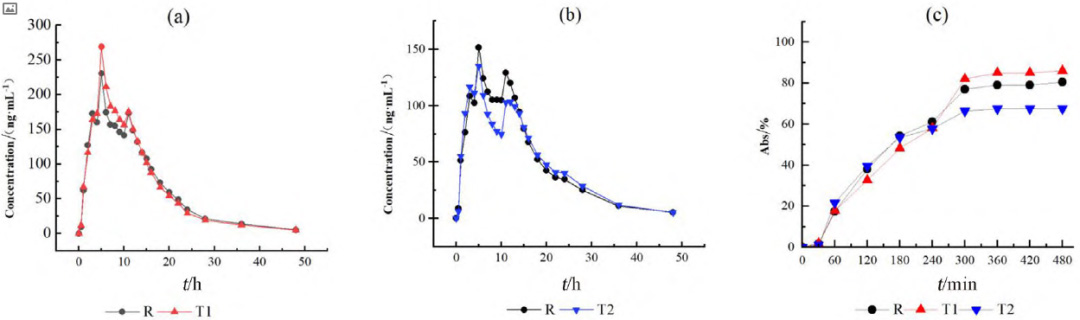
图 1 平均血药浓度 - 时间曲线图(a 和 b)和体内累积吸收百分数图(c)
表 1 T1 和R 的人体试验药动学参数
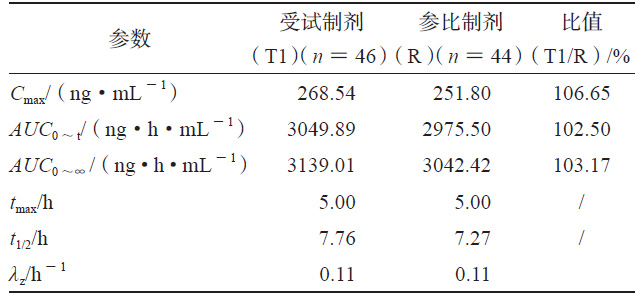
表 2 T2 和R 的人体试验药动学参数
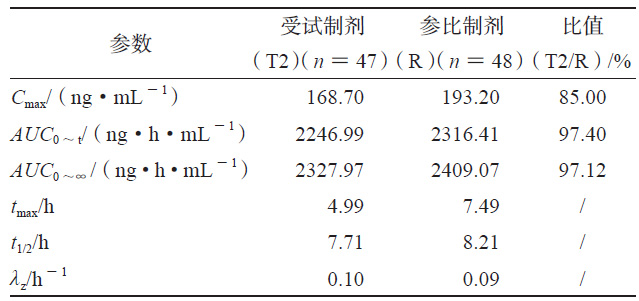
3.2 药物体外溶出试验
3.2.1 药物溶出度测定
由参比制剂体内药动学参数可知,富马酸喹硫平缓释片的体内吸收速率常数Ka约为0.27,3倍吸收半衰期3t1/2约为8h,该数据提示富马酸喹硫平参比制剂8h的累积吸收超过85%。以此为依据,采用流通池法,溶出介质前3h为pH1.2的盐酸溶液,3h后为纯化水介质,流速20 mL·min,温度(37±0.5)℃,分别于0.5、1、2、3、4、5、6、7、8h接取1mL左右的溶出液,进行10 000 r·min,3 min的离心后,取上清液,采用HPLC法进行检测,得到的喹硫平的微分溶出曲线通过数值积分法转换为累积溶出百分数曲线,转换公式如下:
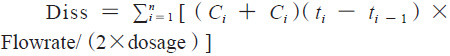
其中,Diss为累积溶出百分数,dosage为药物活性成分总含量,Ci 为ti 时间点的药物浓度,Flowrate为溶出介质流速。
3.2.2 样本检测
使用HPLC检测样品中药物浓度,色谱柱:Agilent 5 TC-C18(2)(150mm×4.6mm);流动相:甲醇-水- 三乙胺(600∶400∶4),调节pH至4.5;检测波长:289nm;流速:1mL·min;柱温:30 ℃;进样量:10μL。
结果R、T1、T2 的8h体外累积溶出曲线和溶出速度具有明显的差异,T1比R的溶出速率快,但T2比R的溶出速率略慢(见图2),该试验结果与各制剂体内试验不同时间的吸收百分数呈相同趋势(见图1c)。
3.2.3 体内外相关性的建立和评估
依据体内外相关性模型计算体内预测累积吸收百分数,再根据卷积分法计算体内预测血药浓度,具体公式如下:
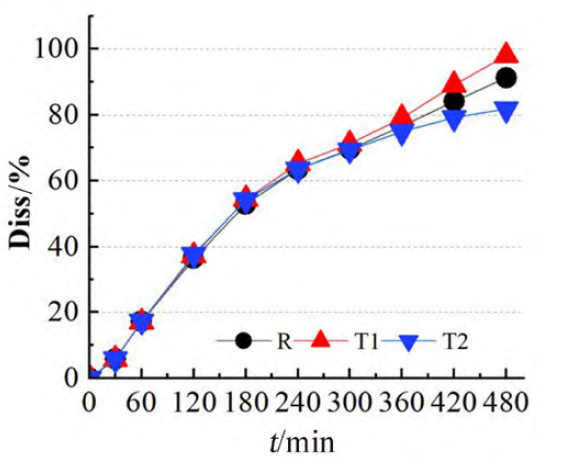
图 2 T1、T2 和R的8h体外累积溶出百分数图(n=6)
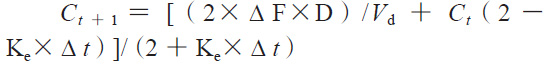
其中,Δ F是两个连续的取样时间点之间的累积溶出分数差,Δ t是两个连续的取样时间点之差,D是药物活性成分总含量,Vd是药物的表观分布容积,Ke是消除速率常数。
采用WinNonlin软件计算累积吸收分数(Abs,%),以三个制剂各时间点的体外累积溶出百分数(Diss,%)为横坐标,以对应时间点的体内累积吸收数据(Abs,%)为纵坐标进行线性分析,可得到T1、T2和R的体内外A级相关性模型(Abs=0.945×Diss+0.337,R2=0.967)。该结果表明该溶出方法具有A级体内外相关性(见图3a)。
使用DDsolver 程序对富马酸喹硫平缓释片的溶出数据进行分析可知其体外溶出特征符合Weibull模型,并得到各制剂累积溶出80%的时间T80,以T80为横坐标,体内实测的AUC0~t为纵坐标进行相关性分析,可得到三个制剂的C级相关图(见图3b)。
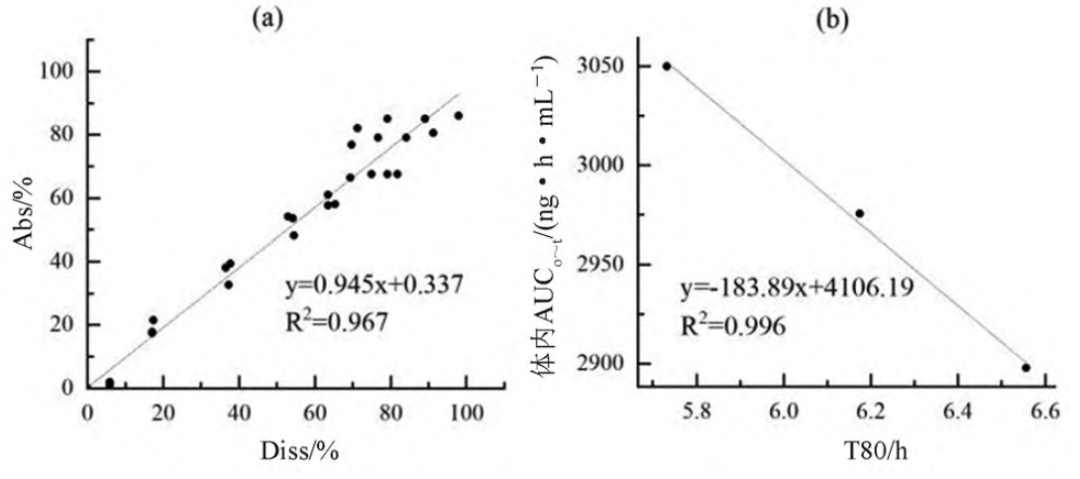
图 3 T1、T2和R的A级(a)及T1、T2和R的C级体内外相关性图(b)
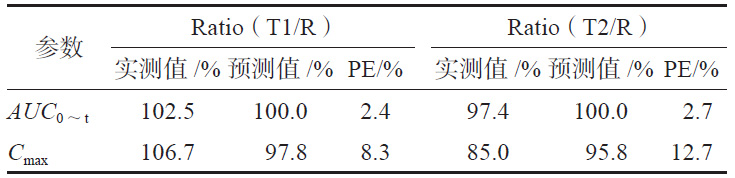
依据模型Abs=0.945×Diss+0.337,R2=0.967,由T1、T2 和R的体外累积溶出百分数可预测三个制剂的体内吸收百分率,由体内吸收百分率通过卷积法可得到制剂的时间-血药浓度数据,进而得到Cmax 和AUC0~t预测的几何均值比Ratio(T/R),并分别计算Ratio(T1/R)和Ratio(T2/R)的预测误差率,T2制剂Cmax的Ratio(T2/R)为12.7%>10%(见表3),需继续进行外部制剂预测能力评估。
表 3 T1、T2 模型预测值与实测值的比较
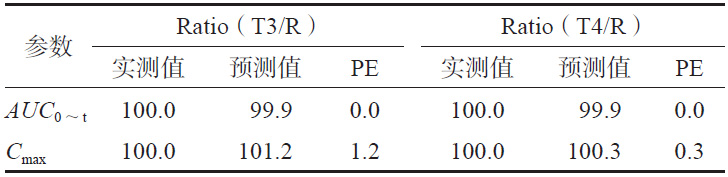
为进一步评估模型的可预测能力,使用市售制剂T3、T4 进行模型外部制剂预测能力评估。依据T1和T2建立的IVIVC模型(Abs=0.945×Diss+0.337,R2=0.967),由制剂T3、T4的体外数据各时间点的Diss(见图4)预测制剂T3、T4 体内各时间点的体内吸收百分率Abs%,进而得到Cmax和AUC0~t预测的几何均值比Ratio(T3/R)、Ratio(T4/R),将市售制剂T3、T4的实测值默认为100%,计算预测误差率PE%,结果见表4。两制剂Cmax 和AUC0~t 预测误差率均小于10%,说明该模型的预测能力良好。
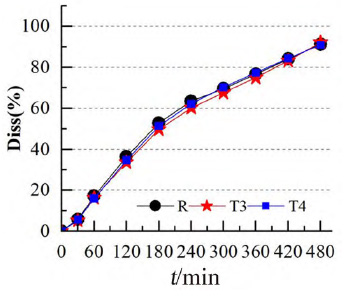
图 4 参比制剂R 和受试制剂T3、T4 的体外累积溶出图(n = 6)
表 4 受试制剂T3、T4 的模型预测值与实测值的比较(%)
04 讨 论
文献报道富马酸喹硫平原料药溶出呈现明显的pH依赖性,溶解度随着pH值的降低而增大,而人体胃肠道存在由酸性胃液到碱性肠液的过渡过程,考虑到胃肠道pH变化可能对药物溶出速率产生的影响,本研究选择了pH 1.2的溶液和纯化水作为溶出介质,并且在3h的时间点进行了介质的转换。相比经典的篮法和桨法,本研究选用的为流通池法,流通池的动力学过程与体内的流体动力学过程更为相似,对于需要进行介质转换的试验,流通池的仪器构造比起篮法和桨法有更多的可操作性。对体内吸收数据的解析也验证了上述设计的必要性,将体内数据和体外数据进行线性拟合后,A级相关和C级相关的拟合优度R2均大于0.9,也说明了该溶出设计的合理性。
本研究的体内试验分为两个独立的试验进行,虽然各试验使用的参比制剂的厂家和批号相同,但在不同的试验中得出的药动学参数的具体数值仍差异较大,如参比制剂R在“T1与R的人体生物等效性试验”中的AUC0~t和Cmax值均比“T2和R的人体生物等效性试验”中的参数值大,为了减少误差,在进行内部预测和外部预测时选用的药动学参数是几何均值比 Ratio(T/R)这类比率值;在进行C级体内外相关性分析时使用的T2制剂的AUC0~t也通过Ratio(T2/R)进行了换算。
目前,文献关于IVIVC研究偏重于其所建立的溶出方法与体内数据的相关性,以线性相关方程和拟合优度R2作为IVIVC研究的结果指标。实际上,IVIVC模型作为一种预测性的模型,其预测能力的评估也是IVIVC研究的重点,FDA建议对于三种释放速率不同的制剂建立的IVIVC模型至少应进行建模制剂内部预测能力的评估,对于两种释放速率不同的制剂进行模型外部制剂预测能力的评估。本研究以三种释放速率不同的制剂建立IVIVC模型,进行内部制剂评估后发现T2制剂Cmax的Ratio(T2/R)的预测误差为12.7%,略微超出10%的评价标准,进一步用外部制剂T3和T4对模型进行了评估,外部制剂预测误差均小于10%,符合评价标准,说明模型具有良好的预测性。
05 结 论
本研究依托流通池法创建富马酸喹硫平缓释片体内外相关性良好的体外溶出方法,对于吸收速率不同的制剂均具有良好的区分力,可为富马酸喹硫平缓释片质量标准的建立提供依据。建立的IVIVC模型经内部验证和外部验证显示其具有良好的可预测性,可通过富马酸喹硫平缓释片制剂的体外特征预测制剂的体内特征,在制剂评价、处方优化等方面都有重要的应用价值。
06 参考文献
略






